

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdfFactor VIII Deficiency
Hemophilia
(Gale Group)
philia A or B. Once such a spontaneous genetic mutation takes place, offspring of the affected person can inherit the newly-created, flawed chromosome.
Demographics
Hemophilia A affects between one in 5,000 to one in 10,000 males in most populations.
One recent study estimated the prevalence of hemophilia was 13.4 cases per 100,000 U.S. males (10.5 hemophilia A and 2.9 hemophilia B). By race/ethnicity, the prevalence was 13.2 cases/100,000 among white, 11.0 among African-American, and 11.5 among Hispanic males.
Signs and symptoms
In the case of severe hemophilia, the first bleeding event usually occurs prior to eighteen months of age. In some babies, hemophilia is suspected immediately, when a routine circumcision (removal of the foreskin of the penis) results in unusually heavy bleeding. Toddlers are at particular risk, because they fall frequently, and may bleed into the soft tissue of their arms and legs. These small bleeds result in bruising and noticeable lumps, but don’t usually need treatment. As a child becomes more active, bleeding may occur into the muscles; a much more painful and debilitating problem. These muscle bleeds result in pain and pressure on the nerves in the area of the bleed. Damage to nerves can cause numbness and decreased ability to use the injured limb.
Some of the most problematic and frequent bleeds occur into the joints, particularly into the knees and elbows. Repeated bleeding into joints can result in scarring within the joints and permanent deformities. Individuals may develop arthritis in joints that have suffered continued irritation from the presence of blood.
Mouth injuries can result in compression of the airway, and, therefore, can be life-threatening. A blow to the head, which might be totally insignificant in a normal individual, can result in bleeding into the skull and brain. Because the skull has no room for expansion, the hemophiliac individual is at risk for brain damage due to blood taking up space and exerting pressure on the delicate brain tissue.
People with hemophilia are at very high risk of hemorrhage (severe, heavy, uncontrollable bleeding) from injuries such as motor vehicle accidents and also from surgery.
Some other rare clotting disorders such as Von Willebrand disease present similar symptoms but are not usually called hemophilia.
Diagnosis
Various tests are available to measure, under very carefully controlled conditions, the length of time it takes to produce certain components of the final fibrin clot. Tests called assays can also determine the percentage of factors VIII and IX present compared to normal percentages. This information can help in demonstrating the type of hemophilia present, as well as the severity.
Individuals with a family history of hemophilia may benefit from genetic counseling before deciding to have a baby. Families with a positive history of hemophilia can also have tests done during a pregnancy to determine whether the fetus is a hemophiliac. The test called chorionic villus sampling examines proteins for the defects that lead to hemophilia. This test, which is associated with a 1% risk of miscarriage, can be performed at 10–12 weeks. The test called amniocentesis examines the DNA of fetal cells shed into the amniotic fluid for genetic mutations. Amniocentesis, which is associated with a one in 200 risk of miscarriage, is performed at 16–18 weeks gestation.
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
525 |
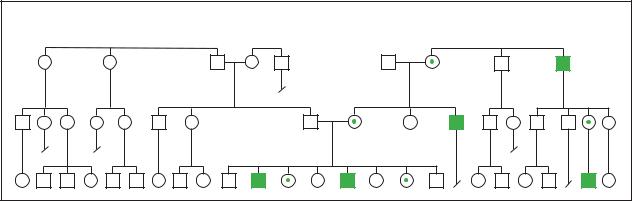
Hemophilia |
Hemophilia A |
|
(Gale Group)
Treatment and management
The most important thing that individuals with hemophilia can do to prevent complications of his disease is to avoid injury. Those individuals who require dental work or any surgery may need to be pre-treated with an infusion of factor VIII to avoid hemorrhage. Also, hemophiliacs should be vaccinated against hepatitis. Medications or drugs that promote bleeding, such as aspirin, should be avoided.
Various types of factors VIII and IX are available to replace a patient’s missing factors. These are administered intravenously (directly into the patient’s veins by needle). These factor preparations may be obtained from a single donor, by pooling the donations of as many as thousands of donors, or by laboratory creation through highly advanced genetic techniques.
The frequency of treatment with factors depends on the severity of the individual patient’s disease. Patients with relatively mild disease will only require treatment in the event of injury, or to prepare for scheduled surgical or dental procedures. Patients with more severe disease will require regular treatment to avoid spontaneous bleeding.
While appropriate treatment of hemophilia can both decrease suffering and be life-saving, complications associated with treatment can also be quite serious. About 20% of all patients with hemophilia A begin to produce chemicals in their bodies which rapidly destroy infused factor VIII. The presence of such a chemical may greatly hamper efforts to prevent or stop a major hemorrhage.
Individuals who receive factor prepared from pooled donor blood are at risk for serious infections that may be passed through blood. Hepatitis, a severe and potentially fatal viral liver infection, may be contracted from pooled factor preparations. Recently, a good deal of concern has
been raised about the possibility of hemophiliacs contracting a fatal slow virus infection of the brain (Creutzfeldt-Jakob disease) from blood products. Unfortunately, pooled factor preparations in the early 1980s were contaminated with human immunodeficiency virus (HIV), the virus which causes AIDS. A large number of hemophiliacs were infected with HIV and some statistics show that HIV is still the leading cause of death among hemophiliacs. Currently, careful methods of donor testing, as well as methods of inactivating viruses present in donated blood, have greatly lowered this risk.
The most exciting new treatments currently being researched involve efforts to transfer new genes to hemophiliacs. These new genes would have the ability to produce the missing factors. As yet, these techniques are not being performed on humans, but there is great hope that eventually this type of gene therapy will be available.
Prognosis
Prognosis is very difficult to generalize. Because there are so many variations in the severity of hemophilia, and because much of what befalls a hemophiliac patient will depend on issues such as physical activity level and accidental injuries, statistics on prognosis are not generally available.
Resources
BOOKS
Genetics and Public Health in the 21st Century: Using Genetic Information to Improve Health and Prevent Disease.
Edited by Muin J. Khoury, Wylie Burke, and Elizabeth J. Thomson. New York: Oxford University Press, 2000.
Hemophilia. Edited by C.D. Forbes, L.M. Aledort, and R. Madhok. New York: Chapman & Hall, 1997.
Resnick, Susan. Blood Saga: Hemophilia, AIDS, and the Survival of a Community. Berkeley: University of California Press, 1999.
526 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

PERIODICALS
Soucie, J.M., et al. “Hemophilia Surveillance System Project Investigators: Occurrence of Hemophilia in the United States.” American Journal of Hematology 59 (1998): 288 .
Stephenson, J. “New Therapies Show Promise for Patients with Leukemia, Hemophilia, and Heart Disease.” JAMA 285 (January 1, 2001): 153 .
ORGANIZATIONS
National Hemophilia Foundation. 116 West 32nd St., 11th Floor, New York, NY 10001. (800) 42-HANDI.http://www.info@hemophilia.org .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
March of Dimes. www.modimes.org .
National Organization for Rare Disorders.www.rarediseases.org .
Jennifer F. Wilson, MS
Hepatocellular carcinoma see Liver cancer
Hepatorenal glycogenosis see Fanconi-
Bickel syndrome
I Hereditary angioneurotic edema
Definition
Hereditary angioneurotic edema (HANE) is a non- sex-linked (autosomal) dominant disease that results from mutations in a gene responsible for producing one of the proteins responsible for human immunity. This disease is also known as hereditary angioedema (HAE) or hereditary C1 inhibitor deficiency because it is a deficiency of the protein (C1-INH) that inhibits the action of the enzyme known as C1 which causes this disease.
Description
There are two recognized forms of HANE. Type I represents approximately 80-85% of the cases of hereditary angioneurotic edema. In this type, the protein C1INH is not produced in sufficient quantities. Type II HANE represents the remaining 15-20% of cases. In this type, C1-INH concentrations are normal, but the C1-INH protein produced is defective.
Related to the two types of hereditary angioneurotic edema are acquired types of this disease (AANE or AAE) that are not based on a defective gene. Type I AAE is caused by a disorder that causes over-growth (proliferation) of the lymph tissues and destroys C1-INH. Type II AAE is caused by the presence of autoantibodies (antibodies that attack the host organism that produced them) that destroy C1-INH. Both of these acquired forms of angioedema can generally be differentiated from the two types of HANE by the age of onset. Symptoms of the acquired diseases usually do not occur until the fourth decade of life, while those of the hereditary forms are generally present prior to puberty.
The human body has two distinct immune systems: the humoral immune system and the cell-mediated immune system. The complement system is a part of the humoral immune system. Humoral means within the humor, or fluids, of the body. Blood, lymph, and bile compose the fluids of the humor. The complement system uses at least 30 different proteins to “mark” any foreign cells in the body that do not have certain protective proteins on their cell membranes which identify them as belonging in the body. These complement proteins are designated C1, C2, C3, et cetera. Once the foreign cells have been “marked,” a particular form of white blood cell, called a phagocyte, is dispatched to the area with the marked cells and destroys them.
Phagocytes will eventually destroy any cell that is marked by complement; therefore, it is important to make sure that the complement proteins are not marking non-foreign cells. When cells are improperly marked, these cells will also be destroyed, causing what is called an autoimmune response. In effect, this autoimmune response means that the body is recognizing itself as foreign and attempting to destroy healthy cells. Inhibitors of the various complement proteins are necessary to prevent these proteins from marking the wrong cells or from continuing to mark cells after the foreign cells have been destroyed.
C1 inhibitor (C1-INH) is a chemical that is involved in the regulation of the complement system by inhibiting the action of the first complement protein (C1). C1-INH acts by binding free C1 molecules in the humor, preventing them from being able to function. It also limits the activation of other complement proteins.
Because C1-INH is diminished or defective in people affected with HANE, C1 is not inhibited and this inappropriately initiates the complement reaction which causes the swelling (acute inflammatory response) characteristic of HANE.
C1-INH also binds to the chemicals kallikrein and plasmin that are involved in blood clotting. Kallikrein is
edema angioneurotic Hereditary
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
527 |

Hereditary angioneurotic edema
K E Y T E R M S
Acquired angioneurotic edema—Abbreviated AANE, or AAE, this is a non-hereditary form of angioedema that generally begins to show symptoms in, or after, the fourth decade of life.
Androgens—A group of steroid hormones that stimulate the development of male sex organs and male secondary sexual characteristics.
Angioneurotic edema—Recurrent episodes of swelling of the tissues of the body caused by an over-active immune system. This is also called angioedema.
C1 inhibitor—Abbreviated C1-INH, this protein is responsible for preventing the action of the C1 complement molecules in the body. It is this protein that is either deficient or malformed in HANE.
Complement system—Class III MHC (major histocompatobility complex) proteins capable of destroying invading organisms directly via natural immunity, as well as indirectly through an interaction with other components of the immune system.
Hereditary angioneurotic edema—Abbreviated HANE, or HAE, this is an inherited kind of angioneurotic edema. Type I HANE is caused by a deficiency of C1-INH. Type II HANE is caused by a malformation of the C1-INH protein.
Kallikrein—A protein necessary for the activation of chemicals that cause dilation of blood vessels to allow increased blood flow to an area that requires more blood than normal. It is also capable of cleaving the complement, C5, into C5a, a much more robust and active form of this complement molecule.
Phagocyte—White blood cells capable of engulfing and destroying foreign antigen or organisms in the fluids of the body.
Plasmin—The blood protein that is responsible for dissolving blood clots.
Urticaria—Also known as hives. Usually associated with an allergic reaction.
necessary for the activation of chemicals that cause dilation of blood vessels to allow increased blood flow to an area that requires more blood than normal. Plasmin is the chemical responsible for dissolving blood clots. A lack of binding of plasmin means that the formation of initial
blood clots is difficult, a problem that is exacerbated by high levels of unbound kallikrein, which allows higher than normal blood flow.
With the absence or dysfunction of the C1-INH protein, the functions of blood flow, blood clotting, and immune response are impaired in individuals affected by hereditary angioneurotic edema, leading to swelling of the bodily tissues.
Genetic profile
The central Pyncheon family in Nathaniel Hawthorne’s The House of the Seven Gables carries an ancestral curse of dying from choking on their own blood. Hawthorne describes members of the family who made odd sounds in the throat and chest when agitated, and sometimes died from choking: “This mode of death has been an idiosyncrasy with his family, for generations past....[the] prophecy was probably founded on a knowledge of this physical predisposition in the Pyncheon race.” It seems possible that Hawthorne was not only describing the symptoms of HANE but also acknowledging it to be an inherited genetic disorder.
All hereditary forms of HANE are caused by mutations in the gene responsible for the production of C1INH. This gene is located on the long arm (q) of chromosome 11, at the specific location q11.2-q13. There are at least 13 different mutations of the C1-INH gene that cause the symptoms of HANE. Six of these are known to cause type I HANE, while another six are known to cause type II HANE. The final mutation has only been found in one individual. In this case, an acquired form of angioedema was determined to be caused by a mutation in a different region of the C1-INH gene than those mutations causing type I or type II cases of HANE.
Demographics
HANE affects approximately 50,000 people in the United States and Europe. It is estimated to occur in approximately one in every 50,000 to 150,000 live births. HANE appears to affect males and females equally and does not have a racial preference.
As an autosomal dominant trait, only one copy of an abnormal gene needs to be inherited for an individual to be affected. Therefore, if one child is affected with HANE, the likelihood that a second child will be affected with HANE is 50%. In cases of parents related by blood (consanguineous parents) the likelihood of HANE is increased.
528 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
Signs and symptoms
Individuals affected with either form of HANE have episodes of swelling of the hands, feet, trunk, face, digestive tract, and airways (angioneurotic edema or angioedema). These attacks of angioedema are often accompanied by attacks of nausea, vomiting, and abdominal pain. The frequency and severity of these attacks is not predictable and varies from individual to individual. These attacks may occur without cause, or they may be triggered by anxiety, stress, or minor traumas, such as dental procedures. If these symptoms are accompanied by hives (urticaria) a diagnosis other than HANE is indicated.
Symptoms of HANE generally first occur prior to puberty and episodes generally increase in severity after puberty.
Diagnosis
A diagnosis of HANE is suspected in individuals who have recurrent attacks of swollen tissues (angioedema). Diagnosis of type I HANE is confirmed by blood tests showing abnormally low levels of C1-INH, C2, and C4. Diagnosis of type II HANE is confirmed by blood tests showing normal levels of C1-INH and C2, but abnormally low levels of C4. Abnormally low levels of C1-INH and C4 without the presence of autoantibodies suggest a diagnosis of type I acquired angioedema, while abnormally low levels of C1-INH and C4 and the presence of autoantibodies suggest a diagnosis of type II acquired angioedema.
Hives (urticaria) are not generally associated with HANE. If hives are present with tissue swelling, this may suggest an allergic reaction, not a case of HANE. Occasionally, individuals affected with HANE also develop hives, but they are usually secondary to the angioedema. In a severe allergic reaction, hives are generally prominent as the major symptom.
Treatment and management
The treatment of both hereditary forms of angioedema is the same. Androgens (male sex hormones) such as winstrol, danazol, and oxandrolone have been shown to be effective in preventing chronic recurrences of swelling. These drugs are seldom used to treat acute attacks. In instances of abdominal attacks, fluid replacement therapy via intravenous injection may be required. Demerol and Compazine suppositories are often prescribed to relieve abdominal pain and vomiting.
Edema (swelling) of the airways is the most lifethreatening feature of HANE. Without prompt medical
attention, individuals affected with HANE can die from an obstruction of the airway caused by this swelling. Unfortunately, if the attending physician does not recognize HANE, attempts at tracheal intubation (formation of an airway directly in the neck) may aggravate the swelling rather than produce a functioning airway.
Treatment with vapor-heated C1-INH concentrate has proven to be an effective treatment both as a prophylactic (preventative) and a treatment for acute attacks of angioedema in all individuals affected with HANE. The C1-INH concentrate is derived from human blood plasma; therefore it may possibly be contaminated. It is vapor-heated to inactivate possible hepatitis and HIV viruses. However, because HANE is a disease of the immune system, many doctors are reluctant to use C1-INH from other people and many patients are unwilling to accept such a treatment. The use of human recombinant C1-INH should alleviate any concerns arising from possible contamination of the blood supply.
Androgens are still the preventative treatment of choice because they are more cost-effective than treatments with C1-INH. However, androgens should not be given to women who are pregnant, or who might become pregnant. In these cases, C1-INH treatment is required.
In 1999, the U.S. Food and Drug Administration granted Orphan Drug Designations to human recombinant C1-INH for both preventative and acute treatment of HANE. On March 21, 2000, Baxter Healthcare’s Hyland Immuno division and Europe’s Pharming Group announced an agreement to jointly develop recombinant human C1-INH. As of the March 2000 press release by these two companies, pre-clinical (animal) studies were expected to be completed in late 2000 and phase I human trials were slated to begin in late 2000 or early 2001. Because of the Orphan Drug Designations from the USFDA, this possible treatment for HANE is automatically “fast-tracked,” which means that it could potentially be approved for human use by 2004.
Prognosis
The key to successful management of HANE is a proper medical diagnosis. With proper medical treatment, HANE is completely controllable and individuals affected with HANE suffer no diminishment in quality of life.
Resources
BOOKS
Hawthorne, Nathaniel. The House of the Seven Gables. New York, New York: Signet Classics Penguin Books Ltd., 1961.
edema angioneurotic Hereditary
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
529 |

Hereditary colorectal cancer
PERIODICALS
Asghar, S., and M. Pasch. “Therapeutic inhibition of the complement system.” Frontiers in Bioscience (September 2000): E63-81.
Cicardi, M., et al. “Pathogenetic and clinical aspects of C1 inhibitor deficiency.” Immunobiology (August 1998): 366376.
Markovic, S., D. Inwards, A. Evangelos, and R. Phyliky. “Acquired C1 esterase inhibitor deficiency.” Annals of Internal Medicine (January 2000): 144-150.
Waytes, A., F. Rosen, and M. Frank. “Treatment of hereditary angioedema with a vapor-heated C1 inhibitor concentrate.” New England Journal of Medicine (June 1996): 1630-1634.
ORGANIZATIONS
Hereditary Angioedema Association. PO Box 492, Live Oak, FL 32064. http://www.hereditaryangioedema.com .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
“Angioedema (Hereditary).” eMedicine. http://www.emedicine
.com/derm/topic24.htm (February 23, 2001). “Angioedema, Hereditary; HAE.” Online Mendelian Inheri-
tance in Man. http://www.ncbi.nlm.nih.gov/htbin-post/ Omim/dispmim?106100 (February 23, 2001).
The Complement Laboratory at the University of Iowa.http://ictg.uiowa.edu/clab/what.htm (February 23, 2001).
OTHER
“Pharming and Baxter to co-develop human C1 inhibitor to treat hereditary angioedema.” Pharming Group N.V. Press Release (March 21, 2000).
Paul A. Johnson
Hereditary arthro-opthalmopathy see
Stickler syndrome
I Hereditary colorectal cancer
Definition
Hereditary colorectal cancer is cancer of the colon or rectum that develops chiefly as the result of inherited factors.
Description
The colon, or the large intestine, is a long muscular tube that absorbs water from stool and advances the stool towards the rectum. The rectum works in conjunction
with the anus to coordinate the process of defecation. The colon and rectum are jointly referred to as the colorectum.
A neoplasm is a portion of abnormal tissue that grows rapidly and out of control. Cancer is the malignant type of neoplasm. Colorectal cancer is a relatively common and dangerous cancer. Tumors originate in the mucosa, or inner lining of the colorectum, and grow inwardly. Eventually, the tumor spreads outwardly until it reaches lymph nodes or other organs in the abdomen. Ultimately, cancer cells may detach from the original tumor and spread to distant parts of the body (such as the liver, lungs, bone, and brain) in a process called metastasis.
The development of colorectal cancer is not a random event, but rather arises in a sequential fashion. The first easily detected step is the appearance of adenomatous polyps. Polyps are grossly defined as elevations of a surface. An adenomatous polyp is derived from the glandular elements of the mucosa. A person may have any number of colorectal adenomatous polyps. Eventually, one or more of these polyps may transform into a cancer. The risk of colorectal cancer increases with the number of polyps. Larger polyps are also more likely to become cancerous than smaller ones. The factors that initiate this adenoma-cancer sequence are inherited and/or acquired from the environment.
Colorectal cancer occurs in certain families much more often than expected by chance alone. In fact, an important and common risk factor for the development of colorectal cancer is the occurrence of colorectal cancer in the family. About 10% of people have a first-degree relative with colorectal cancer. Having a first-degree relative with colorectal cancer increases the chance of developing colorectal cancer by twoto three-fold. The risk becomes even higher when colorectal cancer occurs in a relative at an early age (before 50 years of age) or when more than one relative has the cancer. This suggests that susceptibility of developing colorectal cancer in affected families is due to inherited factors, although shared exposure to environmental stimuli may play a role. Scientists are investigating the genetic factors that may be responsible for the increased risk of colorectal cancer in these cases of common inheritance.
The vast majority of cases of colorectal cancer are sporadic; that is, they occur in the absence of a hereditary syndrome, although familial risk may be involved. But rarely, colorectal cancer is inherited as part of a welldefined syndrome. These syndromes altogether account for about 2-5% of all cases of colorectal cancer.
Familial adenomatous polpyposis
In the syndrome of familial adenomatous polyposis (FAP), adenomas develop in the colon and rectum
530 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
early in life, at an average age of 15 years. Eventually, hundreds to thousands of adenomas will develop. The presence of such a large number of adenomas ensures that at least one of these adenomas will develop into cancer if the colon is not surgically removed. In people with FAP, the average age of occurrence of colorectal cancer is 39. Some patients will develop cancer in their teens and almost every patient will have cancer by age 45.
Other types of polyps are also common in patients with FAP. Polyps may develop in the stomach or duodenum. Those in the stomach are benign, while those in the duodenum may become malignant. The cancer risk in these other polyps is much less than the risk associated with the colorectal polyps. Patients with FAP may also have abnormalities outside the gastrointestinal tract, such as osteomas, desmoid tumors, extra teeth, and hypertrophy of the retinal pigment epithelium.
Three variants of FAP have been identified. Gardner syndrome is a rare variant of FAP characterized by colorectal polyps and a marked prominence of extraintestinal growths. Examples of the growths include osteomas, epidermoid cysts, and desmoid tumors. Although these growths usually present only cosmetic problems, desmoid tumors can occasionally compress nearby tissue in a harmful way.
Turcot syndrome is another rare type of FAP. Patients with this syndrome have the typical colorectal polyps, as well as malignant tumors of the central nervous system such as medulloblastoma, astrocytoma, ependymoma, and glioblastoma multiforme.
Patients with the attenuated adenomatous polyposis coli form of FAP have many colonic polyps, but not the hundreds or thousands seen in typical FAP. The chance of developing colon cancer approaches but does not reach 100%, and colon cancer usually appears later than in patients with typical FAP.
Hereditary nonpolyposis colorectal cancer
Patients with hereditary nonpolyposis colorectal cancer (HNPCC) have about an 80% risk of developing colorectal cancer if untreated. They may have more polyps than the general population, but not the hundreds or thousands of polyps associated with FAP. The average age for the development of cancer is 45 years old. Frequently, a patient with HNPCC will have multiple cancers at the same time (synchronous) or may develop cancers at different time periods (metachronous).
Extraintestinal cancers sometimes occur in HNPCC. The most common is uterine cancer, but other examples include cancer of the uterus, stomach, small intestine, pancreas, kidney, and ovary.
The Amsterdam criteria are clinical criteria for the diagnosis of HNPCC in a family:
•At least three relatives with colorectal cancer, one of whom must be a first-degree relative of the other two.
•Colorectal cancer involving at least two generations.
•One or more cases of colorectal cancer before the age of 50.
Muir-Torre syndrome is a rare form of HNPCC. In addition to polyps and cancer of the colon and rectum, patients exhibit various types of skin cancer.
Genetic profile
It must be understood that all colorectal cancers stem from genetic mutations. Environmental factors may also contribute to the development of cancer. Sometimes colorectal cancer appears in a patient who has neither affected relatives nor an inherited syndrome. Other cases appear in families that seem genetically susceptible to the development of these cancers. The presence of colorectal cancer in relatives, especially young relatives, increases the risk of developing colorectal cancer. In families affected by the rare syndromes of hereditary colorectal cancer (HNPCC, FAP, and their variants), the genetic mutations are inherited in autosomal dominant fashion.
Whether it appears sporadically or is inherited as part of a syndrome, colorectal cancer is generally linked to mutations in certain categories of genes: proto-onco- genes, tumor suppressor genes, DNA mismatch repair genes, or modifier genes. The proto-oncogene category inludes the K-ras, src, and c-myc genes. The tumor suppressor genes are the APC (adenomatous polyposis coli) gene, the DCC (deleted in colon cancer) gene, the MCC (mutated in colon cancer) gene, the DPC4 gene, and p53. The mismatch repair genes are hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6/GTBP. The modifier genes inlude the COX2 (cyclooxygenase 2) gene, the CD44v gene, and the phospholipase A2 gene.
The genetic defect in FAP and its three variants (Gardner syndrome, Turcot syndrome, and attenuated adenomatous polyposis coli) reside on the APC gene, which is on the long arm of chromosome 5. However, there are a wide variety of mutations within the APC gene that can result in those syndromes. Sometimes Turcot’s syndrome is associated with the same mutations as those in HNPCC. Mutations of mismatch repair genes, such as hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6/GTBP, are characteristic of the HNPCC syndrome. The transmission of these hereditary colorectal cancer syndromes occurs through mutations of the same genes that are mutated in sporadic cases of colorectal cancer. But it must be emphasized that the hereditary colorectal cancer
cancer colorectal Hereditary
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
531 |

Hereditary colorectal cancer
K E Y T E R M S
Adenomatous—Derived from glandular structures.
Astrocytoma—Tumor of the central nervous system derived from astrocytes.
Biopsy—The surgical removal and microscopic examination of living tissue for diagnostic purposes.
Central nervous system—In humans, the central nervous system is composed of the brain, the cranial nerves and the spinal cord. It is responsible for the coordination and control of all body activities.
Computed tomography—An imaging procedure that produces a three-dimensional picture of organs or structures inside the body, such as the brain.
Desmoid tumor—Benign, firm mass of scarlike connective tissue.
Distal—Away from the point of origin.
Endoscopy—A slender, tubular optical instrument used as a viewing system for examining an inner part of the body and, with an attached instrument, for biopsy or surgery.
Ependymoma—Tumor of the central nervous system derived from cells that line the central canal of the spinal cord and the ventricles of the brain.
Epidermoid cyst—Benign, cystic tumor derived from epithelial cells.
Glioblastoma multiforme—Tumor of the central nervous system consisting of undifferentiated glial cells.
Medulloblastoma—Tumor of the central nervous system derived from undifferentiated cells of the primitive medullary tube.
Metachronous—Occurring at separate time intervals.
Metastasis—The spreading of cancer from the original site to other locations in the body.
Osteoma—A benign bone tumor.
Polyp—A mass of tissue bulging out from the normal surface of a mucous membrane.
Prophylactic—Preventing disease.
Proximal—Near the point of origin.
Synchronous—Occurring simultaneously.
syndromes are inherited in an autosomal dominant pattern. This means that each child of an affected person has a 50% chance of inheriting the disease.
Families with the inherited syndromes of colorectal cancer can undergo genetic testing to determine which individuals have inherited the disease. The tests for the defective genes can detect the mutation in approximately 60 to 80% of FAP families and about 50% of HNPCC families. However, if one person is found to have the mutation, the other family members can be tested with nearly 100% accuracy. Although genetic testing can provide useful information to the patients, it may be associated with psychosocial risks. Thus, genetic testing should be performed only in formal programs. Genetic counseling should also be provided.
Demographics
Colorectal cancer is relatively common with approximately 160,000 new cases diagnosed each year, but the syndromes of inherited colorectal cancer are rare. It is estimated that they comprise only 2-5% of all cases of colorectal cancer. FAP occurs in about one in every 10,000 births. The incidence of all colorectal cancer increases with age.
Signs and symptoms
The clinical manifestations of colorectal cancer depend largely on location and tumor size. Tumors in the proximal colon can grow to large sizes before detection. They may cause weight loss, abdominal pain, or bleeding. The bleeding may be readily noticed by the patient as frank blood in the toilet, or smears of blood in the stool. Less extensive bleeding may be detected by the fecal occult blood test, in which a sample of stool obtained during a rectal exam is tested for microscopic amounts of blood. Anemia, or low red blood cell count, detected by a laboratory test may prompt further examination of the colon to determine if a tumor is the source of bleeding. In the smaller, distal colon, tumors are more likely to cause obstruction. This may cause gas pains and decrease in the caliber of the stool. Additionally, these cancers may cause bleeding or a change in bowel habits. In FAP, the first symptom is usually diarrhea.
Diagnosis
The presence of symptoms such as abdominal pain, weight loss, change in bowel habits, or decrease in stool caliber may point to a diagnosis of colorectal cancer. Of course, these symptoms must be interpreted within the context of the patient’s age, previous medical history, and family history of colorectal cancer.
532 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
Ideally, the diagnosis of colorectal cancer should be made before symptoms develop. A number of screening tests are useful for detecting colorectal cancer. The fecal occult blood test that was discussed earlier is a simple test performed in the office. The normal result is the absence of blood in the stool. If blood is found in the stool, the suspicion for colorectal cancer becomes higher. Standard screening also includes an endoscopic exam— either sigmoidoscopy or colonoscopy. In these exams, a thin, specially lighted tube is inserted directly into the anus and advanced into the colon. The physician can view the inside of the colon and check for polyps or tumors. Sigmoidoscopy allows examination of the lower part of the colon while colonoscopy allows a more extensive view. Sometimes a barium enema is added to the screening procedure. In this test, a dye is injected into the anus and up into the colon. The dye coats the inside of the colon so that tumors can be detected by plain x ray.
New screening tests are currently under investigation. In wireless endoscopy, a tiny pill-sized camera is swallowed. As the camera traverses the gastrointestinal tract, it transmits video footage that can be examined for suspicious abnormalities. Eventually the camera is passed out of the anus with the stool. Virtual colonoscopy generates a three-dimensional image of the colon by applying advanced computer graphics technology to images obtained by computed tomography (CT) scanning. These processes can spare the patient the usual discomfort of traditional endoscopy. However, they are not yet fully developed nor approved for colorectal cancer screening.
If any of the above screening tests identifies an abnormality that appears to be a tumor, the diagnosis must be confirmed by biopsy. This is performed during colonoscopy. A small piece of tissue is removed and examined in the laboratory for cancerous cells.
Most medical organizations recommend that screening should begin in the general population at age 40 to 50. The fecal occult blood test is performed annually and sigmoidoscopy every three to five years. If a first degree relative has colorectal cancer, then screening should begin at 35 to 40 years of age. Alternatively, screening can begin five years earlier than the age of a young relative who has colorectal cancer.
Individuals in families affected by hereditary colorectal cancer syndromes are at high risk for developing cancer early in life. Therefore, screening is initiated at a young age. Screening can be reserved for those family members who have been proven to carry the abnormal gene by genetic testing, or it can be applied to all family members if the specific mutation cannot be identified. Some experts propose that in families with a history of FAP, screening should begin at 10 to 12 years of age and
be repeated every one to two years. In families with HNPCC, colorectal screening should begin at 20 to 30 years of age and also be repeated every one to two years.
Since FAP and HNPCC are also associated with other cancers, affected patients should undergo appropriate screening for these malignancies as well. Those with FAP require regular upper endoscopy to detect tumors of the stomach and duodenum. Women with HNPCC should undergo screening for uterine cancer by way of random biopsies of the inner lining of the uterus.
Treatment and management
The treatment of sporadic colorectal cancer requires surgical removal of the tumor and surrounding tissue. Chemotherapy or radiation therapy may also be necessary. But the treatment of colorectal cancer in the hereditary syndromes is more aggressive. In these cases, the entire colon must be removed, since cancer will almost certainly develop in any remaining colon. Sometimes the rectum is also removed; alternatively, the patient may undergo frequent examination of the rectum for polyps or cancers. Experts strongly recommend that individuals with known FAP should consider surgical removal of the colon and/or rectum early in life as a prophylactic measure, before cancer is diagnosed. Although the role of prophylactic surgery in patients with HNPCC is less well-defined, many experts favor it. The patient faces a choice between prophylactic surgery and frequent, lifelong screening.
Some studies have shown that the drug sulindac may reduce the number of adenomatous polyps that develop in FAP and its variants. In addition, certain nonsteroidal antiflammatory drugs such as aspirin may also reduce the incidence of colorectal cancer in general.
Prognosis
Patients with a hereditary colorectal cancer syndrome such as FAP, HNPCC, or its variants, have a much higher likelihood of developing colon cancer than the general population. In the extreme case of typical FAP, essentially 100% of patients will develop colon cancer without surgery. If colon cancer does develop, survival depends on the extent to which the cancer has spread. Cancer that is isolated to the colon is associated with much better survival than cancer that has spread to distant organs such as the liver or lungs.
Resources
BOOKS
“Colon and Rectum.” In Sabiston Textbook of Surgery. Edited by Courtney Townsend Jr., et al. 16th ed. Philadelphia: W.B. Saunders Company, 2001.
cancer colorectal Hereditary
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
533 |

Hereditary desmoid disease
“Familial Colon Cancer” and “Predisposition to Colorectal Cancer.” In Sleisenger & Fordtran’s Gastrointestinal and Liver Disease. Edited by Mark Feldman, et al. Sixth ed. Philadelphia: W.B. Saunders Company, 1998.
PERIODICALS
Lynch, Henry and Trudy Shaw. “The Genetics of Colorectal Cancer.” Primary Care & Cancer (June 1999).
Kevin Osbert Hwang, MD
I Hereditary desmoid disease
Definition
Hereditary desmoid disease (HDD) is a condition that causes people to develop a benign (noncancerous) growth known as a desmoid tumor. Desmoid tumors may also be called fibromatosis.
Description
In HDD, multiple family members from several generations develop desmoid tumors. These tumors are very rare. They account for fewer than 0.1% of all tumors diagnosed. The term “desmoid” comes from the Greek word for “band.” That describes these tumors well, as they have a tendonor ligament-like appearance. They usually occur in the abdomen, but they may also develop in the neck, chest, arms, and legs.
Desmoid tumors may appear due to mutations, or changes, in a gene called adenomatous polyposis coli (APC). Most desmoid tumors, though—more than 97%—occur sporadically, meaning that they are not caused by genetic mutations. People who develop sporadic desmoid tumors have no other health problems associated with mutations in the APC gene and have no close family members with the tumors. In the past desmoid tumors were classified as fibrosarcomas (growths associated with cancer), but this is no longer the case.
Mutations in the APC gene usually result in familial adenomatous polyposis (FAP). This condition causes hundreds to thousands of polyps (tiny growths) to develop in the colon. It is associated with a high risk for developing colon cancer. People who have FAP need to have their health monitored on a regular basis. Colon cancer can be prevented by careful medical screening and removal of the colon.
Some families with FAP develop extra-colonic symptoms (involving organs other than the colon), including desmoid tumors. The combination of colon polyposis and desmoid tumor was once termed
“Gardner’s syndrome,” but it is now known that the two conditions are the same. Other extra-colonic features seen in families with FAP are cysts in the jawbone, skin cysts (epidermal cysts), bony bumps on the skull, a specific kind of spot on the retina, and thyroid cancer. About 10% of people with FAP will develop desmoid tumors. However, the risk differs from family to family.
In HDD, multiple family members over two or more generations develop desmoid tumors, but not colon polyposis. Family members in subsequent generations will have an increased risk of developing desmoid tumors.
Genetic profile
Every person diagnosed with HDD has a 50% chance of passing on the condition to each of his/her children. The chances that a child who has the gene mutation associated with HDD will develop a desmoid tumor are thought to be very high, maybe even 100%. It is possible that there may be other genes involved in HDD, but no gene other than APC has been identified. The location of the mutation within the APC gene may predict the symptoms and health problems that a person will experience, but this association is far from perfect.
Demographics
Hereditary desmoid disease is a rare condition. As of 2001, only four families have been reported in the medical literature. (It is likely, however, that not all families with HDD have been described in the literature.) Males and females are equally affected.
Signs and symptoms
Desmoid tumors may cause a noticeable lump and/or pain.
Diagnosis
HDD is usually diagnosed solely upon family history. Evaluation for HDD requires filling out a detailed, three-generation family tree. Medical records and/or death certificates should also be examined to confirm or clarify possible diagnoses of desmoid tumors. Medical records for family members developing colon polyps and/or undergoing colon surgery will also be requested in order to evaluate for FAP.
Genetic (or diagnostic) testing for APC gene mutations (changes) is another way of making a diagnosis. It may be offered to someone who has developed a desmoid tumor and has a family history of such tumors. If a mutation is identified, the positive test result provides proof of the diagnosis. If no mutation is identified, this negative
534 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |