

10. Hydrogen bonding and complex formation |
463 |
TABLE 1. Apparent stability constant Kc of complexes between aromatic amines and 1-fluoro-2,4-dinitrobenzene (unless otherwise indicated) at 40 °C
|
|
|
Kc |
|
|
Amine |
Solvent |
mol 1 dm3 b |
Reference |
||
Aniline |
benzene |
0.068 |
|
|
253 |
Aniline |
chloroform |
0.70 |
|
|
253 |
Anilinea |
benzene |
0.068a |
|
253 |
|
Anilinea |
chloroform |
0.068a |
|
253 |
|
Anilinea,c |
ethanol |
0.34a,c |
(0.49) |
256 |
|
Anilinea,c |
ethanol/ethyl acetated |
0.12a,c |
(0.17) |
256 |
|
Anilinea,c |
ethyl acetate |
0.05a,c |
|
256 |
|
Aniline |
tetrahydrofuran |
0.20 |
(0.31) |
257 |
|
2H-Aniline-d7e |
chloroform |
0.62e |
|
|
253 |
N-Methylaniline |
benzene |
0.66 |
|
|
253 |
N-Methylaniline |
tetrahydrofuran |
1.06 |
(0.65) |
257 |
|
N,N-Dimethylaniline |
benzene |
0.44 |
|
|
253 |
p-Methoxyaniline |
benzene |
0.67 |
|
|
253 |
p-Methoxyaniline |
tetrahydrofuran |
1.17 |
(1.58) |
257 |
|
p-Methoxyaniline |
chloroform |
0.37 |
|
|
258 |
p-Methoxyaniline |
carbon tetrachloride |
1.5 |
|
|
258 |
p-Methoxyaniline |
chlorobenzene |
0.29 |
|
|
258 |
p-Methoxyaniline |
1,2-dichloroethane |
0.95 |
|
|
258 |
p-Methoxyaniline |
1,4-dioxane |
0.81 |
|
|
258 |
m-Methoxyaniline |
benzene |
0.055 |
|
|
253 |
m-Methoxyaniline |
tetrahydrofuran |
0.20 |
(0.43) |
257 |
|
p-Methylaniline |
benzene |
0.24 |
|
|
253 |
p-Methylaniline |
tetrahydrofuran |
0.41 |
(0.31) |
257 |
|
m-Methylaniline |
benzene |
0.11 |
|
|
253 |
m-Methylaniline |
tetrahydrofuran |
0.43 |
(0.16) |
257 |
|
p-Chloroaniline |
benzene |
0.022 |
|
|
253 |
p-Chloroaniline |
tetrahydrofuran |
0.46 |
(0.29) |
257 |
|
a1-Chloro-2,4-dinitrobenzene.
bCalculated from UV/VIS spectrophotometric analyses by Benesi Hildebrand treatment. Range of values used in the determination: from 410 to 450 nm. In parentheses: values calculated from kinetic data.
c At 23.8 °C.
d50:50, by volume.
eCalculated from 1H NMR data in CDCl3.
and 0.48 (mol 1 dm3) respectively, at 26 °C, with the electron donor partner being the
solvent in all cases254. |
|
|
|
|
|
|
The main consequence of EDA interactions |
between |
solvents |
of high |
donicity |
||
and nitroarenes is that amines |
in benzene (or |
in other |
similar |
solvents) |
compete |
|
with the solvents in complexing the |
nitroarenes253,254. This fact |
explains |
the ratio |
|||
Kcchloroform/Kcbenzene D 10 for |
the |
molecular complex between aniline and FDNB |
||||
(see Table 1).
Table 2 shows some Kc values of equilibria like 29 between nitroarenes and aliphatic amines. Even if differences in the mechanism of interactions (as well as differences in experimental conditions, in particular solvents and temperature) make a full comparison difficult, some main points from data of Table 2 are worthy of consideration.
(i) The introduction of a fluorine atom on the nitro-containing substrate stabilizes the complex: KcFDNB > Kc1,3 DNB as required if the fluoro derivative interacts with amines by hydrogen bonding.

464 |
Luciano Forlani |
TABLE 2. Apparent constant of stability of molecular complexes between nitroaromatic derivatives and aliphatic amines in various solvents
Substratea |
Amine |
Solvent |
T °C |
Kc mol 1 dm3 b |
Reference |
1,2-DNB |
dibutylamine |
n-hexane |
27 |
0.25 |
255 |
1,2-DNB |
tributylamine |
n-hexane |
27 |
0.098 |
255 |
1,3-DNB |
n-butylamine |
n-hexane |
27 |
0.39 |
255 |
1,3-DNB |
diethylamine |
n-hexane |
27 |
0.24 |
255 |
1,3-DNB |
di-n-butylamine |
n-hexane |
27 |
0.20 |
255 |
1,3-DNB |
triethylamine |
n-hexane |
27 |
0.089 |
255 |
1,3-DNB |
tributylamine |
n-hexane |
27 |
0.043 |
255 |
1,4-DNB |
n-butylamine |
n-hexane |
27 |
0.36 |
255 |
1,4-DNB |
dibutylamine |
n-hexane |
27 |
0.17 |
255 |
1,4-DNB |
tributylamine |
n-hexane |
27 |
0.032 |
255 |
FDNB |
n-butylamine |
toluene |
21 |
14c |
259 |
FDNB |
n-butylamine |
cyclohexane |
21 |
27 |
260 |
FDNB |
piperidine |
cyclohexane |
21 |
79 |
260 |
CDNB |
piperidine |
cyclohexane |
21 |
3.4 |
260 |
CDNB |
n-butylamine |
cyclohexane |
21 |
0.08 |
260 |
FDNB |
DABCOd |
benzene |
25 |
0.31; (0.31)c |
261 |
FDNB |
TEe |
benzene |
40 |
0.47c |
258 |
TNB |
DBUf |
toluene |
18 |
12 |
245 |
a 1,2-DNB D 1,2-dinitrobenzene; 1,3-DNB D 1,3-dinitrobenzene; 1,4-DNB D 1,4-dinitrobenzene; FDNB D 1-fluoro-
2,4-dinitrobenzene; CDNB |
D |
1-chloro-2,4-dinitrobenzene; TNB |
D |
1,3,5-trinitrobenzene. |
|
||
b |
|
|
|
|
|
||
|
Calculated from UV/VIS spectrophotometric data, unless otherwise indicated. |
|
|||||
c Calculated from kinetic data. |
|
|
|
|
|||
d |
|
|
|
|
|
|
|
e DABCO D 1,4-diaza[2.2.2]bicyclo-octane. |
|
|
|
|
|||
TE D triethylamine. |
|
|
D 7.3 at 25 °C, 5.7 at 33 °C; HŁ |
D 39 kJ mol 1, S D |
|||
f DBU D 1,8-diazabicyclo[5,4,0]undec-7-ene, Kc |
|||||||
357 J mol 1 K 1. |
|
|
|
|
|
|
|
(ii)The stability order KcFDNB > KcCDNB for n-butylamine and piperidine indicates that a hydrogen bond is operating.
(iii)The presence of steric requirements for the formation of complexes is an important point to explain relative stabilities of complexes. The system aliphatic amines/dinitrobenzenes in hexane reveals the presence of an interaction, which is defined
as an electron donor acceptor (EDA) interaction255. The donor ability decreases by passing from primary amines (n-butylamine) to secondary amines (di-n-butylamine, diethylamine) and to tertiary amines (tri-n-butylamine and triethylamine) as indicated by the differences in stability of the complexes. The order Kcprimary > Kcsecondary > Kctertiary is explained by the steric requirements of the electron donor for the formation of the complexes which are usual when donor acceptor interaction is operating.
(iv) Piperidine is more prone to interacting with both FDNB and CDNB than n- butylamine (Kcpiperidine > Kcn-butylamine). Generally, cyclic secondary amines have less steric requirements than non-cyclic secondary amines.
The change in the relative positions of the nitro groups affords the trends 1,2- > 1,3- > 1,4-dinitrobenzene, for the stability of the complexes, but the observed differences are moderate.
Recently262, the apparent stability constants of the complexes between aromatic fluoro derivatives and amines (shown in equilibrium 31, Kc in mol 1 dm3) in toluene-d8 were evaluated by 19F chemical shift measurements.
ArF |
C |
HNRR0 |
Kc |
Ð |
31 |
|
|||||
|
|
ArF HNRR0 |
|||
|
|
|
|
|
|

10. Hydrogen bonding and complex formation |
465 |
The main interaction of the complex of equilibrium 31 is probably a hydrogen bonding interaction between nitrofluorobenzenes and some amines. These complexes are more stable when the nitro groups are in position 2 of fluorobenzene than when they are in position 4. A reasonable explanation of this trend is the interaction of the ortho nitro group with the amine shown in 110, in which a second hydrogen bond (between the amino and the nitro groups) enhances the interaction.
For n-butylamine, the Kc2-nitrofluorobenzene/Kc4-nitrofluorobenzene ratio equals 4, while for the secondary amine piperidine the ratio is higher (Kc2-nitrofluorobenzene/Kc4-nitrofluorobenzene D 9). This supports an interaction between the two nitrogen atoms as shown in 111, where the nitrogen of the amine is the electron donor and the nitrogen of the nitro group is the electron acceptor.
|
R |
|
H |
N |
|
R |
|
|
|
||
H |
H |
H |
N |
F |
O− |
F |
O− |
+N |
O |
|
N O |
|
|
|
+ |
(110) |
(111) |
The self-association of amines (see Section II.C) is a complication which is almost impossible to evaluate, in all the quantitatively studied systems involving protic amines, in associating solvents, including the data and observations reported here.
B. Some Kinetic Features of Aromatic Nucleophilic Substitution Reactions
The usual kinetic law for SNAr reactions is the second-order kinetic law, as required for a bimolecular process. This is generally the case where anionic or neutral nucleophiles react in usual polar solvents (methanol, DMSO, formamide and so on). When nucleophilic aromatic substitutions between nitrohalogenobenzenes (mainly 2,4- dinitrohalogenobenzenes) and neutral nucleophiles (amines) are carried out in poorly polar solvents (benzene, hexane, carbon tetrachloride etc.) ‘anomalous’ kinetic behaviour may be observed263. Under pseudo-monomolecular experimental conditions (in the presence of large excess of nucleophile with respect to the substrate) each run follows a first-order
kinetic law, but the rate constants (kobs in s 1 mol 1 dm3) were not independent of the initial concentration value of the used amine. In apolar solvents the most usual kinetic
feature is the increase of the kobs value on increasing the [amine]0 values; [amine]0 indicates the initial concentration value of the amine.
This kinetic feature is observed by using primary and secondary amines, both aromatic and aliphatic. Tertiary amines263 (and other substances unable to produce substitution products such as 2-hydroxypyridine264) may act as a catalyst: in apolar solvents, in a series of runs carried out without changing the initial concentration value of substrate and of amines, it was found that the addition of tertiary amines enhances kobs values.
In more polar solvents, as reported in 1954 by Ross and Kuntz256, for reactions between 2,4-dinitrochlorobenzene and aniline, kobs values decrease with increasing [amine]0 values. A possible interpretation of this kinetic feature is the formation of a molecular compound between aniline and 2,4-dinitrochlorobenzene by a quickly established equilibrium, which

466 |
Luciano Forlani |
precedes the substitution process. In principle, such complex kinetic features are usually attributed to the presence of some equilibria preceding the reactions and involving the reactants and the solvent265,266.
It is known that when the kinetic behaviour cannot be studied in the absence of the molecular complex (and the reactivity of the free reagent is unknown), the kinetic law by itself cannot indicate whether the complex observed is on the reaction pathway or is a non-productive equilibrium267. In SNAr reactions discussed here, the usual experimental conditions [ArF]0 < [RNH2]0 are used. In some cases, the kobs values obtained under experimental conditions [ArF]0 > [RNH2]0 may be assumed to involve only the uncatalysed process. As a consequence, it is possible to say that the presence of complexes (in apolar solvent) clearly produces an enhancement of the reactivity and a positive catalysis, while in polar solvents256 the presence of complexes depresses the rate of substitution.
Scheme 14 has long been considered263,268 the explanation of this kinetic behaviour with the intervention of a second molecule of catalyst (or of the same reacting amine) on the zwitterionic intermediate to promote the departure of the proton and of the leaving group.
+
NHRR′
|
k1 |
− |
k2 |
|
ArL + HNRR′ |
|
Ar |
ArNRR′ + HL |
|
|
|
k −1
L k3
catalyst
SCHEME 14
The more popular mechanism of HL elimination263 (where L is the leaving group) is the SBGA mechanism (Specific Base General Acid) which involves a base-catalysed abstraction of the proton and the acid catalysed departure of the leaving group from the zwitterionic intermediate 112 (where G are electron-withdrawing groups) of the usual twostep mechanism of SNAr reactions. In some cases, a concerted mechanism of departure of the proton and the leaving group is operating.
N
H H
+
L N
−
G
(112)
On the basis of the presence of the molecular complexes discussed in the previous section and of other observations253,260, Scheme 15 was proposed as an alternative to the more usually accepted mechanism shown by Scheme 14. The left part of Scheme 15 is the uncatalysed reaction pathway, while the part on the right is the catalysed one.

10. Hydrogen bonding and complex formation |
467 |
|||||||||
|
|
|
Catalyst (HNRR′) |
|
|
|
|
|
||
ArL |
|
Kc |
|
Molecular Complex |
|
|||||
|
|
|
|
|
||||||
|
|
|
|
|||||||
|
|
+ HNRR′ |
|
+ HNRR′ |
|
|||||
k°−1 k°1 |
|
k1 k−1 |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
zwitterionic |
|
zwitterionic |
|
||||||
|
|
|
|
|||||||
|
intermediate |
|
intermediate |
|
|
|||||
|
|
|
k°2 |
|
|
|
k2 |
|
|
|
Products
SCHEME 15
Equation 32 is derived from Scheme 15 and allows the evaluation of Kc values reported (in parentheses) in Tables 1 and 2.
kobs 1 C Kc[RNH2]0 D k0 C Kck[RNH2]0 |
32 |
By considering k D k1/k 1 k2 and k0 D k10/k 10 k20, the first condition to observe catalytic behaviour is that k > k0. Obviously when k0 D k, no effect of the variation in the initial amount of amine may be detected and kobs is an actual second-order rate constant; when k0 < k, a decrease in kobs values on increasing [RNH2]0 values may be observed.
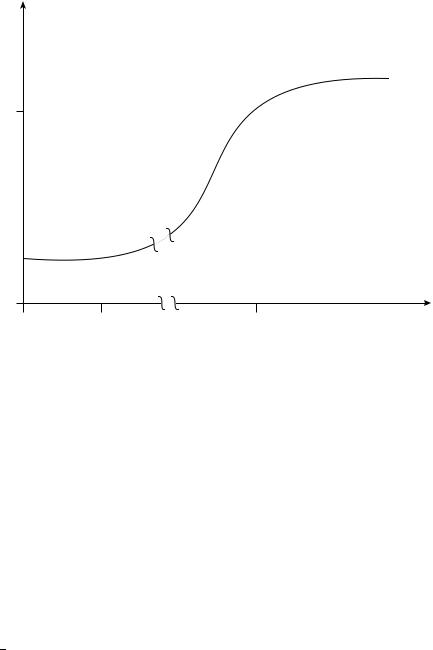
When Kc[RNH2]0is − 1, k0 (for low values of initial amine concentration when fluoro derivatives are used, or when Kc is a low value as in cases of chloro derivatives), kobs is k0 (the uncatalysed process). When Kc[RNH2] becomes a high value (because [RNH2] is increased), there is a leveling-off of the rate as shown in Figure 1.
The general overall kinetic behaviour of Figure 1 may be observed (under the experimental conditions [ArF]0 < [RNH2]0) if a large range of concentration values of amine is used, as in the case of the reactions between 2,4-dinitro-fluorobenzene and n-butylamine in benzene269 or toluene259. The major part of cases reported in the literature concerns a more restricted range of [amine]0 values.
The first horizontal part of the plot of Figure 1 (A) corresponds to the substitution process in the absence of catalysis, kobs D k0. Consequently, it allows the evaluation of the reactivity of the ‘free’ substrate. This value is confirmed by runs carried out under experimental conditions [ArF]0 > [RNH2]0 (which are unusual in these investigations): the same kobs value is obtained, in agreement with a second-order kinetic law, for reaction of ‘free’ substrate.
The second part (B) of the plot is referred to the presence of both processes: the substitution reaction of the ‘free’ substrate and the reaction of the complexed substrate. The plateau (C) is a leveling-off of the rate and corresponds to the saturation of the catalytic process: in this range of [RNH2]0 values the main reaction is that of the complexed substrate.
A very interesting (and important) detail of the SNAr reaction showing positive catalysis is the dependence of catalysed and uncatalysed processes on the temperature and was investigated in several instances270 274. There are systems271,272 in which the experimental reaction rate constant (in s 1 mol 1 dm3) is decreased on increasing the temperature. In other instances the increase in the temperature doesn’t have an effect on kobs273.
468 |
Luciano Forlani |
kobs
(s−1 mol−1dm3)
(C)
0.5
(B)
(A)
[RNH2]0 (mol dm−3)
0
0 |
0.002 |
0.2 |
FIGURE 1. Diagram to show the effect of the increase in the initial concentration value of amine on the rate of substitution expressed as a second-order rate coefficient. Indicative [RNH2]0 values and kobs values are from reactions between 2,4-dinitrofluorobenzene and n-butylamine in toluene259
In the case of the reactions between 1,3,5-trinitrobenzene (TNB) and 1,8- diazabicyclo[5,4,0]undec-7-ene (DBU)245, in toluene, shown in Scheme 16, regarding the formation of a zwitterionic -complex, kobs values are increased on increasing the [DBU]0 values: there is a catalytic effect of the base in a system without leaving group and proton. The increase in kobs values is related to the presence of the molecular complex between TNB and DBU indicated by Scheme 16.
Analysis of the data allows the observation that the substrate/nucleophile association (Kc values, see Table 2) is clearly decreased on increasing the temperature, while the k1 and k10 of Scheme 16 (the attack of the DBU on the ‘free’ TNB and on the complexed TNB, respectively) are increased on increasing the temperature, showing activation parameters usual in SNAr reactions.
The reaction of 1-fluoro-2,4,6-trinitrobenzene and 2,4-dimethoxyaniline, in cyclohexane, shows a negative activation enthalpy274 ( 30 kJ mol 1), in agreement with a ‘desolvative association mechanism’ in which the nucleophile competes with the solvent in associating with the substrate in an equilibrium preceding the substitution process.
The reactions 33 between tetrachloro-N-n-butylphthalimide (113) and n-butylamine275 in aprotic and apolar media (cyclohexane, benzene, toluene, xylenes) show a third experimental reaction order in the amines explained by the formation of a complex (n -like) between the electron acceptor substrate (the derivative of the phthalimide) and the electron donor nucleophile (the amine). In mixed solvents (such as the mixtures cyclohexane/aromatic solvents) the kinetic investigation reveals the presence of a competition between the electron donor solvent and the amine in complexing the substrate.

10. Hydrogen bonding and complex formation |
469 |
|||||
TNB |
+ DBU |
Kc |
|
[molecular complex] |
|
|
|
|
|
||||
|
|
|
||||
|
k°1 k°−1 |
|
k −1 |
+ DBU |
|
|
|
|
k1 |
|
|||
|
|
|
|
N |
|
|
|
|
|
+ |
|
|
|
|
|
H |
|
N |
|
|
|
O2 N |
|
NO2 |
|
|
|
|
|
|
− |
|
|
|
|
|
|
NO2 |
|
|
|
|
SCHEME 16 |
|
|
|||
Cl |
O |
|
|
|
|
|
|
|
|
|
|
||
Cl |
C |
|
|
|
|
|
|
N(CH2 )3 CH3 + H2 N(CH2 )3 CH3 |
|
||||
Cl |
C |
|
|
|
|
|
O |
|
|
|
|
||
Cl |
|
|
|
|
||
|
|
|
|
|
|
|
(113) |
|
|
|
|
|
(33) |
|
|
|
|
Cl |
O |
|
|
|
Cl |
|
|
C |
|
|
|
|
|
N(CH2 )3 CH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N(CH2 )3 CH3 |
|
|
|
Cl |
|
|
C |
|
|
|
|
|
Cl |
O |
|
Another reaction mechanism explaining the observed enhancement of kobs values with increasing [amine]0 values in SNAr reactions is the ‘dimer mechanism’276, which involves the self-association of the amines277 279 and which (in some cases) may be considered overlapped with mechanism of Scheme 14. A reaction pathway for ‘dimer mechanism’ is shown in Scheme 17. Considering the zwitterionic intermediate 114 it is possible to have a catalysis in removing the proton and the leaving group (reaction pathway indicated
by k3).
On the other hand, it is reasonable to admit that several different mechanisms may be in competition to afford the products of the reaction. Under particular experimental conditions, a mechanism may predominate over another with a specific reaction pathway;
470 |
|
|
Luciano Forlani |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
R |
N ..... |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
H |
H . |
H |
|
|
|
|||
|
|
|
|
|
|
|
|
|||||
|
|
|
.. |
|
|
|
|
|||||
|
|
|
|
|
|
|
|
. |
+NHR |
|
|
|
|
|
|
|
|
|
k1 |
|
. L |
k2 |
|
|
|
O2 N |
L + RN |
H |
|
NR |
|
|
NO2 |
Products |
||||
|
|
|
|
|||||||||
|
k−1 |
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
||
|
H |
|
|
|
|
|
|
− |
|
k3 |
|
|
|
NO2 |
|
|
|
|
|
|
|
|
Catalyst |
|
|
NO2
(114)
SCHEME 17
in other cases, depending on the experimental conditions, another reaction pathway may be the one leading to the final product.
Anyway, it is important to remember that a mechanism may hardly be demonstrated: a mechanism is a scientific theory which may be proved false by further observations280. In principle, our data are only able to give us some indications to the possible pathways of the reactions.
VII. REFERENCES
1.C. Reichardt, Solvent Effects in Organic Chemistry, Verlag Chemie, New York, 1979.
2.J. E. Leffler and E. Grunwald, Rates and Equilibria of Organic Reactions, Wiley, New York, 1963.
3.V. Gutmann, The Donor Acceptor Approach to Molecular Interaction, Plenum Press, New York, 1978.
4.E. Grunwald and S. Winstein, J. Am. Chem. Soc., 70, 846 (1948).
5.E. N. Kosower, J. Am. Chem. Soc., 78, 5700 (1956).
E. N. Kosower, J. Am. Chem. Soc., 80, 3253 (1958).
E. N. Kosower, J. Am. Chem. Soc., 80, 3261 (1958).
6.K. Dimroth, C. Reichardt, T. Siepmann and F. Bohlmann, Ann. Chem., 661, 1 (1963).
7.C. Reichardt, Chem. Rev., 94, 2319 (1994).
8.V. Gutmann, Coordination Chemistry in Non-Aqueous Solvents, Springer, Wien, 1968.
9.U. Maier, V. Gutmann and W. Gerger, Monatsh. Chem., 106, 1235 (1975).
10.R. Zahradnik and P. Hobza, Pure Appl. Chem., 80, 245 (1988).
11.S. Leutwyler, U. Even and J. Jortner, J. Chem. Phys., 79, 5769 (1983); S. Leutwyler, J. Chem. Phys., 81, 5480 (1984).
12. P. Hobza and R. Zahradnik, Intermolecular Complexes, Chap. I, Elsevier, Amsterdam, 1988,
p. 25.
13.J. Elquero, C. Marzin, A. R. Katritzky and P. Linda, The Tautomerism of Heterocycles, Academic Press, London, 1976.
14.L. Forlani, J. Heterocycl. Chem., 29, 1461 (1992); M. Annese, L. Forlani, A. C. Bonamartini, C. Rizzoli and P. Sgarabotto, J. Chem. Soc., Perkin Trans. 2, 615 (1994).
15.P. Gilli, V. Ferretti, V. Bertolasi and G. Gilli, in Advances in Molecular Structure Research (Eds. M. Hargittai and I. Hargittai), Vol. 2, JAI Press Inc., Greenwich, 1995.
16.R. Huisgen and H. Waltz, Chem. Ber., 89, 2616 (1956); M. Davies and D. K. Thomas, J. Org. Chem., 60, 767 (1956); M. E. Hobbs and W. W. Bates, J. Am. Chem. Soc., 74, 746 (1952).
17.M. H. Kracov, C. M. Lee and H. G. Mautner, J. Am. Chem. Soc., 87, 892 (1965).
18.M. Mascal, Contrib. Org. Synthesis, 1, 31 (1994).
19.J. S. Nowick and J. S. Chen, J. Am. Chem. Soc., 114, 1107 (1992).
20.A. Eschenmoser and E. Loewenthal, Chem. Soc. Rev., 21, 1 (1992).
10. Hydrogen bonding and complex formation |
471 |
21.W. H. Pirkle and T. C. Pochapsky, Chem. Rev., 89, 347 (1989).
22.W. H. Pirkle, T. C. Pochapsky, G. S. Mahler, D. E. Corey, D. S. Reno and D. M. Alessi, J. Org. Chem., 51, 4991 (1986).
23.L. Pauling, The Nature of the Chemical Bond, Cornell Press, New York, 1959.
24.G. C. Pimentel and A. L. McClellan, The Hydrogen Bond, W. H. Freeman Co., San Francisco, 1960.
25.S. E. Schullery and R. M. Scott, J. Chem. Soc., Perkin Trans. 2, 1559 (1984).
26.M. H. Abraham, Chem. Soc. Rev., 22, 73 (1993).
27.G. A. Jeffrey and W. Soenger, Hydrogen Bonding in Biological Molecules, Springer-Verlag, Berlin, 1991.
28.G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989.
29.C. B. Aakeroy¨ and K. R. Seddon, Chem. Soc. Rev., 22, 397 (1993).
30.P. Hobza and R. Zahradnik, Intermolecular Complexes, Chap. V, Elsevier, Amsterdam, 1988.
31.F. Vogtle,¨ Supramolecular Chemistry, Wiley, New York, 1991.
32.J. M. Lehn, Angew. Chem., Int. Ed. Engl., 27, 90 (1988).
33.J. M. Lehn, Angew. Chem., Int. Ed. Engl., 29, 1304 (1990).
34.J. S. Lindsey, New J. Chem., 15, 153 (1991).
35.S. J. Geib, C. Vincent, E. Fan and A. D. Hamilton, Angew. Chem., Int. Ed. Engl., 32, 119 (1993).
36.J. Emsley, Chem. Soc. Rev., 9, 91 (1968).
37.A. C. Legon, Chem. Soc. Rev., 22, 153 (1993).
38.A. C. Legon and D. J. Millen, Chem. Soc. Rev., 21, 71 (1992).
39.M. C. Etter, Acc. Chem. Res., 23, 120 (1990).
40.M. H. Abraham, J. Phys. Org. Chem., 6, 660 (1993).
41.M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin
Trans. 2, 521 (1990); M. H. Abraham, P. L. Grellier, D. V. Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 699 (1989).
42. M. H. Abraham, G. J. Buist, P. L. Grellier, R. A. McGill, D. V. Prior, S. Oliver, E. Turner, J. J. Morris, P. T. Taylor, P. Nicolet, P. C. Maria, J. F. Gal, J. L. M. Abboud, R. M. Doherty,
M. J. Kamlet, W. J. Shuely and R. W. Taft, J. Phys. Org. Chem., 2, 540 (1989).
43.J. Li, Y. Zhang, H. Ouyang and P. W. Carr, J. Am. Chem. Soc., 114, 9813 (1992).
44.M. Meot-Ner (Mautner) and S. C. Smith, J. Am. Chem. Soc., 113, 862 (1991).
45.C. T. Seto and G. M. Whitesides, J. Am. Chem. Soc., 113, 712 (1991).
46.C. A. Hunter, Chem. Soc. Rev., 23, 101 (1994).
47. G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. N. Chin, M. Mammen and
D. M. Gordon, Acc. Chem. Res., 28, 37 (1995).
48.J. A. Zerkowski and G. M. Whitesides, J. Am. Chem. Soc., 114, 5473 (1992).
49.J. P. Mathias, E. E. Simanek, J. A. Zerkowski, C. T. Seto, and G. M. Whitesides, J. Am. Chem. Soc., 116, 4316 (1994).
50. E. E. Simanek, M. Mammen, D. M. Gordon, D. Chin, J. P. Mathias, C. T. Seto and
G. M. Whitesides, Tetrahedron, 51, 607 (1995).
51.J. P. Mathias, E. E. Simanek, C. T. Seto and G. M. Whitesides, Angew. Chem., Int. Ed. Engl., 32, 1766 (1993).
52.J. P. Mathias, C. T. Seto, E. E. Simanek and G. M. Whitesides, J. Am. Chem. Soc., 116, 1725 (1994).
53.J. P. Mathias, E. E. Simanek and G. M. Whitesides, J. Am. Chem. Soc., 116, 4326 (1994).
54.P. Barczynski, Z. Dega-Szafran and M. Szafran, J. Chem. Soc., Perkin Trans. 2, 765 (1985).
55.J. E. Barry, M. Finkelstein and S. D. Ross, J. Org. Chem., 49, 1669 (1984).
56.M. M. Davies in The Chemistry of Nonaqueous Solvents (Ed. J. J. Lagowski), Vol 3, Academic Press, London, 1970.
57.M. Ilczyszyn, J. Chem. Soc., Faraday Trans., 90, 1411 (1994).
58.S. Rozycka´-Roszak and E. Turska, J. Chem. Soc., Perkin Trans. 2, 227 (1985).
59.Z. Dega-Szafran and M. Szafran, J. Chem. Soc., Perkin Trans. 2, 897 (1987).
60.D. D. Perrin, Dissociation Constants of Organic Bases in Aqueous Solution, Butterworths, London, 1965.
61.M. Orban,´ A. Kiss and L. Barcza, J. Chem. Soc., Perkin Trans. 2, 1815 (1987).
62.A. Buvari´ and L. Barcza, J. Chem. Soc., Perkin Trans. 2, 543 (1988).
63.T. Gramstad and L. J. Stangeland, Acta Chem. Scand., 47, 605 (1993).
64.T. Gramstad, Acta Chem. Scand., 46, 1087 (1992).
472 |
Luciano Forlani |
65.T. Gramstad, Acta Chem. Scand., 47, 985 (1993).
66.G. Gonzalez´ and E. Clavijo, J. Chem. Soc. Perkin Trans. 2, 1751 (1985).
67.Y. Wang, H. Li, J. Chao, F. Zhang and M. Dai, Wuli Huaxue Xuebao, 217 (1994); Chem. Abstr., 121, 82412q (1994).
68.N. Stahl and W. P. Jencks, J. Am. Chem. Soc., 108, 4196 (1986).
69.M. J. Tubergen and R. L. Kuczkowski, J. Am. Chem. Soc., 115, 9263 (1993).
70.J. Reuben, J. Am. Chem. Soc., 109, 316 (1987).
71.U. Berg and W. P. Jencks, J. Am. Chem. Soc., 113, 6997 (1991).
72.R. Nakagaki, I. Aoyama, K. Shimizu and M. Akagi, J. Phys. Org. Chem., 6, 261 (1993).
73.A. Gavezzotti and G. Filippini, J. Phys. Chem., 98, 4831 (1994).
74.R. Taylor and O. Kennard, Acc. Chem. Res., 17, 320 (1984).
75.R. Taylor, O. Kennard and W. Versichel, J. Am. Chem. Soc., 105, 5761 (1983).
76.R. Taylor, O. Kennard and W. Versichel, Acta Crystallogr., Sect. B, B40, 280 (1984).
77.R. Taylor, O. Kennard and W. Versichel, J. Am. Chem. Soc., 106, 244 (1984).
78.T. W. Panunto, Z. Urbanczyk´-Lipkowska, R. Johnson and M. C. Etter, J. Am. Chem. Soc., 109, 7786 (1987).
79.J. Masnovi, R. J. Baker, R. L. Towns and Z. Chen, J. Org. Chem., 56, 176 (1991).
80.M. J. Kamlet, J. L. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 48, 2877, (1983).
81.L. Giacomelli, R. Cattana, J. Anunziata, J. J. Silber, M. Hedrera, A. Salerno and I. Perillo, J. Phys. Org. Chem., 7, 162 (1994).
82.C. N. R. Rao and T. Pradeep, Chem. Soc. Rev., 20, 477 (1991).
83.P. Hobza and R. Zahradnik, Chem. Rev., 88, 871 (1988).
84.H. Landeck, H. Wolff and R. Gotz, J. Phys. Chem., 81, 718 (1977).
85.D. M. Brewis, N. B. Chapman, J. S. Paine, J. Shorter, and D. J. Wright, J. Chem. Soc., Perkin Trans. 2, 1787 (1974).
G.Pannetier, L. Abello, Bull. Soc. Chim. France, 1645 (1966).
86.J. C. Schug and W. M. Chang, J. Phys. Chem., 75, 938 (1971).
87.N. S. Nudelman, J. Phys. Org. Chem., 2, 1 (1991).
88.A. Arcoria, E. Maccarone, G. Musumarra and G. A. Tomaselli, Tetrahedron, 31, 2523 (1975).
89.J. H. Lady and K. B. Whetsel, J. Phys. Chem., 68, 1001 (1964).
H.Landeck, H. Wolff and R. Gotz,¨ J. Phys. Chem., 81, 718 (1977).
H. Wolff and R. Wurtz, J. Phys. Chem., 74, 1600 (1970).
90.J. L. Mateos, R. Cetina and O. Chao, J. Chem. Soc., Chem. Commun., 519 (1965).
91.M. Kern, B. Servais, L. Abello and G. Pannetier, Bull. Soc. Chim. France, 2763 (1968).
92.H. Wolff and G. Gamer, J. Phys. Chem., 76, 871, (1972).
93.J. H. Lady and K. B. Whetsel, J. Phys. Chem., 69, 1596 (1965).
94.K. B. Whetsel and J. H. Lady, J. Phys. Chem., 68, 1010 (1964).
95.R. L. Werner, J. M. Quinn and J. K. Haken, Spectrochim. Acta, 38A, 887 (1982).
96.J. E. Sinsheimer and A. M. Keuhnelian, Anal. Chem., 46, 89 (1974).
97.L. K. Vinson and J. J. Dannenberg, J. Am. Chem. Soc., 111, 2777 (1989).
98.S. Martinez-Carrera, Acta Crystallogr., 20, 783 (1966).
99.P. Goldstein, J. Ladell and G. Abowitz, Acta Crystallogr. Sect. B, 25, 135 (1969).
100.F. Ramondo, L. Bencivenni, G. Portalone and A. Domenicano, Struct. Chem., 5, 1 (1994).
101.H. A. Staab and T. Saupe, Angew. Chem., Int. Ed. Engl., 27, 865 (1988).
102.R. W. Alder, Tetrahedron, 46, 683, (1990).
103. P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 116, 909 (1994); G. Gilli,
V. Bertolasi, V. Ferretti and P. Gilli, Acta Crystallogr., B49, 564 (1993).
104.F. Hibbert, Acc. Chem. Res., 17, 115 (1984).
105.M. Eigen, Angew. Chem., Int. Ed. Engl., 3, 1 (1964).
106.A. L. Llamas-Saiz, C. Foces-Foces and J. Elguero, J. Mol. Struct., 328, 297 (1994).
107.R. D. Chambers, S. R. Korn and G. Sandford, J. Fluorine Chem., 69, 103 (1994).
108.B. Brzezinski, G. Schroeder, E. Grech, Z. Malaraski and L. Sobczyk, J. Mol. Struct., 274, 75, (1992).
109.B. Brzezinski, E. Grech, Z. Malaraski and L. Sobczyk, Polish J. Chem., 68, 261 (1994).
110.B. Brzezinski, E. Grech, Z. Malaraski and L. Sobczyk, J. Chem. Soc., Perkin Trans. 2, 1267 (1991).
111.B. Brzezinski, E. Grech, Z. Malaraski and L. Sobczyk, J. Chem. Soc., Perkin Trans. 2, 857 (1991).