

8. Complex formation involving double-bonded functional groups |
407 |
Non-cyclic amidines (120 of Scheme 44) react as ambident nucleophiles in the reactions with TNB in DMSO forming198 the two zwitterionic intermediates 121 and 122 of Scheme 44, when the nucleophilic centre is the nitrogen or the carbon atom, respectively. 121 and 122 are in equilibrium with unprotonated forms. Some 122 -complexes were isolated as crystalline solids. In 122, a further nucleophilic attack is carried out by the CDNH group and this forms a meta bridged compound (123) in protonated or unprotonated forms198.
The reaction of Scheme 44 is an interesting synthetic route for preparing heterobicyclic ring systems199 such as 124, 125, 126.
N
N
N N
N N
(124)(125) (126)
N-Salicylideneaniline (127 and 128) derivatives present some interesting aspects200 of the tautomeric process of equilibrium 19. The position of equilibrium 19 depends on the presence of an internal hydrogen bond201,202.
OH |
O H |
N |
N |
|
(19) |
(127) |
(128) |
N,N0-Bis(salicylidene)-p-phenylenediamine (129 and 130; see equation 20) in the crystalline state shows203 a short intramolecular hydrogen bond. The molecules are planar and form a one-dimensional column. The interplanar space is shorter than in other salicylideneanilines; this fact is an indication that there is an intermolecular CT complex.
O H
N |
N |
|
H O |
|
(129) |
(20)
O H
N N
H O
(130)
408 |
Luciano Forlani |
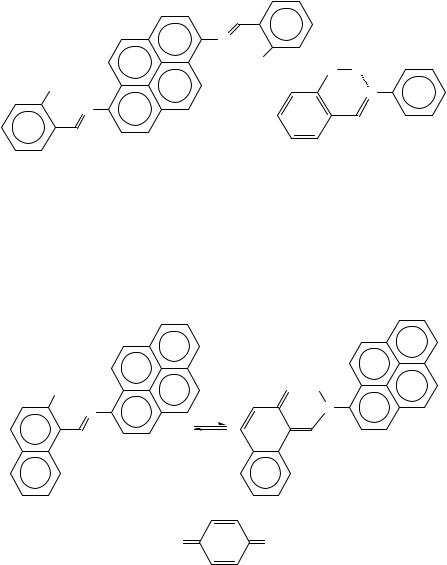
Optical measurements of N,N0-disalicylidene-1,6-pyrenediamine (131) indicate204 the absence of a thermal proton transfer, but the crystals of 129 and 130 are thermochromic; spectroscopic changes with changes of temperature are caused by the transfer of proton from the OH group to the imine nitrogen (equilibrium 20). The keto-form (130) includes electron-donor and electron-acceptor parts: they interact in an intermolecular CT complex203, in the crystalline state. Similar behaviour is reported for the N- tetrachlorosalicylideneaniline and tetrachlorosalicylidene-1-pyrenylamine pair205.
N
HO
O H
OH
N
N
(131) |
(132) |
The hydrogen bond (the NÐ Ð ÐH O hydrogen bond is short) is responsible for the formation of a pseudoaromatic ring (see 132) which is planar206. Geometrical properties of the moieties used and the presence of substituents can modify the pseudoaromatic hydrogen bond. Thus, the position of the hydrogen is sensitive to the presence of CT complexes, as observed in the case of some complexes207 between N-(2-hydroxy-1-naphthylmethylene)- 1-pyreneamine (equilibrium 21) and 7,7,8,8-tetracyanoquinodimethane (133) derivatives. Probably, the modification of charge distribution (caused by the formation of CT complexes or by changes of substituents on aromatic rings208) in 132 and in other similar compounds is an important parameter in stabilizing the pseudoaromatic hydrogen bond.
OH |
|
O H |
|
|
|
|
N |
N |
|
|
(21)
(CN)2 C |
C(CN)2 |
(133)
8. Complex formation involving double-bonded functional groups |
409 |
VI. COVALENT COMPLEXES INVOLVING C=O AND C=N GROUPS AND
NUCLEOPHILES
In principle, when carbonyl, thiocarbonyl or iminoyl groups react with a nucleophile, a covalent bond is formed and the carbon hybridization changes from sp2 to sp3 (equilibrium 22).
X −
C X + Nu |
|
|
|
|
|
|
Nu+ |
(22) |
C |
|
|||||||
|
|
|||||||
|
|
|||||||
|
|
|||||||
(X = O, S, NR)
Water addition to the carbonyl group of aldehydes and ketones has been known for a long time. ˛-Halogenoketones react with water to form tetrahedral adducts (reaction 23).
|
|
O |
|
|
|
OH |
|
|||||||
|
|
|
|
|
|
CHCl2 + H2 O |
|
|
|
|
|
|
|
(23) |
Ph |
|
C |
|
|
Ph |
|
C |
|
CHCl2 |
|||||
|
|
|
|
|||||||||||
|
|
|
|
|||||||||||
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
|
||||||
Kinetic data on equilibrium 23 indicate209 that in mixed solvents a large number of protons participate in the TS of hydration (134) until it reaches the minimum value of 2 (135).
H |
|
H |
H |
H |
O |
O |
n |
O |
O |
Ph |
|
|
Ph |
|
C |
H |
|
C |
H |
O |
|
|
O |
|
CHCl2 |
H |
|
CHCl2 |
H |
|
|
|
||
(134) |
|
|
(135) |
|
The attack of the nucleophile that forms anionic adducts (or zwitterionic adducts, with neutral nucleophiles) parallels the other attacks of nucleophiles on derivatives bearing unsaturated (sp2) carbon atoms, such as alkenes or aromatic substrates, which are both activated by electron-withdrawing groups. The formation of anionic (or zwitterionic) complexes between nitroaromatic derivatives and nucleophiles is a well-known step in nucleophilic aromatic substitution reactions155,210.
The constant of equilibrium 22 may be calculated by several methods. A competitive method was used211 to evalute the equilibrium constant in the formation of cyanohydrin anions of substituted benzaldehydes in DMSO (equilibrium 24).
|
|
O− |
|
||||
ArCHO + CN − |
K |
ArC |
|
|
|
H |
(24) |
|
|||||||
|
|
|
|
||||
|
|
|
|
||||
|
|
||||||
|
|
|
|
|
|
|
|
|
|
CN |
|
||||
(136)
410 |
Luciano Forlani |
The source of cyanide ions is the Meisenheimer adduct formation between trinitrobenzene and cyanide ions in equilibrium 25. The constant of this equilibrium 25 is well known; if the equilibrium concentration of free cyanide ions is negligible, the concentration of the strong red 137 provides a proof of equilibrium 24. Also, in the case of equilibrium 24, DMSO enhances the reactivity of the anionic nucleophile compared with protic solvents.
|
|
|
H |
CN |
|
|
|
|
|
NO2 |
|
O2 N |
NO2 |
|
O2 N |
|
|
|
|
|
|||
|
+ |
CN |
− |
− |
(25) |
|
|
|
|
||
|
NO2 |
|
|
NO2 |
|
|
|
|
|
(137) |
|
The addition of nucleophiles to the carbonyl group may be catalysed by acids obtained by the protonation of the carbonyl oxygen (equilibrium 26). Acid catalysis can also occur during the elimination step which follows the addition step. For example, the reactions of aldehydes with amines (and of all the ammonia derivatives) to form imines are generally assumed to occur in two steps: the first is the addition of nucleophile to yield a gem amino alcohol, the second includes the elimination of water from the tetrahedral adduct 138 (see Scheme 45). This elimination is usually thought to be catalysed by electrophiles171,212.
C |
O |
|
+ H + |
|
|
+ |
+ |
|
|
|
|
|||||||
|
|
|
|
|
C OH |
|
|
|
|
|
C |
|
OH |
(26) |
||||
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
OH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
H |
|
|||||||||||
|
|
|
|
|
|
|
||||||||||||
C O + |
|
|
|
|
|
|
C N |
|
− H2 O |
|
||||||||
|
N |
|
|
|
|
|
|
|
|
|
|
|
C |
N |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
H |
|
|
H |
|
|
H |
|
|
|
|
|
|
|
H |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(138) |
|
|
|
|
|
|
|
|
|
|
|
||
SCHEME 45
Depending on the experimental conditions either the addition or the elimination step may be rate determining.
The formation of the tetrahedral intermediate 138 may be preceded by the zwitterionic complex 139, as the first step of the nucleophilic attack on the partially positive carbon of the carbonyl group.
O− H
C N+ H
H
(139)
While Scheme 45 is generally accepted, only very few direct observations of 1,1-amino alcohols (138) or of the zwitterionic intermediate 139 were reported213.
8. Complex formation involving double-bonded functional groups |
411 |
Direct inspection (by 1H NMR) of mixtures of 4-nitrobenzaldehyde, or pyridine-4- carbaldehyde, in hexadeutero DMSO and piperidine reveals the presence of 140 in the reaction mixtures214.
The starting aldehyde H˛ signal (υ D 10.36) was shifted to υ D 5.33 as required for a change of sp2 to sp3 hybridization by passing from 4-nitrobenzaldehyde to derivative 140 (equation 27). Similar behaviour was observed for mixtures of aromatic aldehydes and of some primary amines (butylamine, t-butylamine): the presence of the 1,1- aminoalcohol (141) was observed for short times, after the imine (142)215 had been formed (equation 28).
H α |
|
OH |
|
|
|
C O + HN |
O2 N |
C N |
|
|
|
|
|
|
|
|
H α |
(27) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(140) |
|
|
O2 N |
|
|
|
|
|
|
|
|
|
|
|
|
OH |
|
H |
|
|||
|
|
|
|
|
|
|
|
|
|
ArCHO + H2 NC4 H9 |
|
|
|
|
|
|
− H2 O |
|
|
|
|
|
|
|
|
|
|||
Ar |
|
C |
|
NHC4 H9 |
|
C NC4 H9 |
(28) |
||
|
|
|
|||||||
|
|
|
|||||||
|
|
|
|
|
|
|
|
Ar |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|||||
|
|
|
|
|
|
||||
|
(141) |
|
(142) |
|
|||||
Equilibrium 29 regarding the tautomerism of pentane-2,4-dione (143) was found to have been completely shifted towards the enol form by a tertiary amine. The predominance of the enol form can be attributed to strong hydrogen-bonding interactions with the amine as shown in 145 and 146. Low polarity of solvents also favours the enol form 144.
CH3 |
CH2 |
CH3 |
|
CH3 |
CH |
CH3 |
C |
|
C |
|
C |
|
C |
|
|
|
||||
O |
|
O |
|
O |
|
(29) |
|
|
|
O |
|||
|
|
|
|
|
H |
|
|
(143) |
|
|
|
(144) |
|
The interaction shown in 146 was investigated by X-ray diffraction216 of the adduct between diethylamine and 143. This dimeric complex (147) arises mainly from hydrogenbonding interactions and shows a three-centred hydrogen bond.
The interactions between the carbonyl carbon of 143 and triethylamine shown in 148, 149 and 150 appear217 to be a better explanation than the reported (and usually accepted) hydrogen-bonding interactions of 143 in triethylamine. IR and 1H-NMR investigations217 show the presence of a labile adduct between 143 and triethylamine which reduces theCH2 1H-NMR signal of the keto form. By using an excess of 143 or reducing the temperature or diluting the 143/triethylamine mixtures with CDCl3, there is an enhancement of the CH2 signal of the keto form.
The use of tributylamine or of diisopropylethylamine favours the keto form, as expected if a nucleophilic interaction of the amine on a CDO carbon atom is the driving force for enolization217. This conclusion is supported also by ab initio calculations217 on the
412 |
|
|
|
|
Luciano Forlani |
|
|
|
|
||
|
CH3 |
CH |
|
CH3 |
|
CH3 |
|
CH |
CH3 |
|
|
|
|
|
|
|
|
||||||
|
C |
C |
|
|
|
|
|||||
|
|
|
|
|
C |
|
C |
|
|||
|
|
|
|
|
|
|
|
− |
|
||
|
O |
O |
|
|
|
|
|
|
|
||
|
|
|
|
|
O |
|
O |
|
|||
|
|
|
|
H |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NEt3 |
|
|
|
+ NEt3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(145) |
|
|
|
|
|
|
(146) |
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
|
|
|
|
C |
CH |
|
|
|
|
|
|
|
Et2 N |
H |
O |
C |
CH3 |
|
|
|
|
|
O |
H |
|
|
|
H |
O |
|
|
|
|
CH3 |
C |
|
O |
H |
NEt2 |
|
|
|
|
|
|
|
CH |
C |
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
|
|
|
|
(147) |
|
|
|
|
|
|
CH3 |
+NEt3 |
CH3 |
CH3 |
+ NEt3 CH |
C |
CH3 |
+NEt3 CH |
CH3 |
|||
CH2 |
|
|
C |
|
|
CH3 |
C |
||||
|
C |
C |
|
|
|
|
|
|
|
C |
|
|
|
|
|
|
|
|
|
|
|
||
|
O_ |
O |
|
|
O |
− |
O |
|
|
O |
OH |
|
|
|
|
H |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
(148) |
|
|
|
|
(149) |
|
|
(150) |
|
|
|
|
|
|
|
|
|
NH3 |
|
|
|
|
|
|
|
|
H |
|
CH |
|
H |
|
|
|
C C
O O
H
(151)
malonodialdehyde/NH3 system: the carbon/nitrogen interaction (151) presents an energy state that is lower than those for the more usual hydrogen-bonding interactions.
1,1,1-Trifluoroacetylacetone (152) reacts by a condensation reaction 30 with aromatic aldehydes218 in the presence of piperidine and acetic acid, in benzene. Triethylamine does not catalyse this reaction. Probably, the product 153 is obtained via the carbinolamine (154), which in turn yields the carbonyl group of the aldehyde. Obviously, the tertiary amine cannot displace a proton and the interaction forms a zwitterionic adduct (see 148).
CH3COCH2COCF3 |
C ArCHO ! ArCHDCHCOCH2COCF3 |
30 |
(152) |
(153) |
|

8. Complex formation involving double-bonded functional groups |
413 |
|||||||||||
NC5H10 |
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
OH |
|
|
|||||
CH |
|
CF3 |
|
|
|
+ |
|
|
|
|||
CF3 |
|
|
CH3 |
|
C |
|
PEt3 |
|
|
|||
C |
C |
|
|
|
Br− |
|
||||||
|
|
|
|
|
|
|
|
|
|
|||
OH |
OH |
|
|
|
|
|
CH3 |
|
|
|||
(154) |
|
|
|
|
(155) |
|
|
|
|
|||
1-Hydroxyphosphonium salts and 1-(trimethylsiloxy)phosphonium salts are obtained219 from the addition of small-sized phosphines (PMe3 and PEt3) to a carbonyl group of ketones and aldehydes, in the presence of chlorotrimethylsilane or of the mixture acetone/bromine, which is a source of anhydrous HBr.
X-ray diffraction provides a structural analysis of the adduct obtained between acetone and triethylphosphine (155) in the presence of bromine. The process of obtaining 155 is an acid-catalysed process, while the adducts obtained in the presence of chlorotrimethylsilane may be ascribed to neutral attack of the nucleophile leading to the zwitterionic complex, as shown in Scheme 46. In Scheme 46 triethylphosphine may be substituted by trimethylphosphine. When triphenylphosphine or tributylphosphine were used, no reactions were observed, owing to the reduced nucleophilic power of the bulky phosphines and to the insolubility of the obtained adducts.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R1 |
|
|
|
|
|
|
|
|
|
R1 |
|
||||||||||||
|
R2 |
|
|
|
|
|
|
X− |
PEt3 |
|
|
R2 |
|
|
|
|
|
|
X− |
||||||||
C |
|
OH |
C |
|
OH |
||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||
|
|
|
|
||||||||||||||||||||||||
+ HX |
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
R1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
PEt3 |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
C O |
|
|
|
|
|
|
|
− HX |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
R2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
+ PEt3 |
|
R1 |
|
|
|
|
|
|
|
|
|
R1 |
|
||||||||||||||
|
R2 |
|
|
|
|
|
O− |
|
Me3SiCl |
|
|
R2 |
|
|
|
|
|
|
|
|
|
Cl− |
|||||
|
|
C |
|
|
|
|
C |
|
OSiMe3 |
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
+ |
|
PEt3 |
|
|
|
|
|
|
|
|
+ |
|
PEt3 |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
R1 = H, Me; R2 = Me
R1 = H,; R2 = Ph
SCHEME 46
Usually, the reactions of carbonyl compounds and derivatives of ammonia are considered to be concluded with the formation of the imino derivative (156), but there is evidence that the CDN double bond may react faster than the CDO group with nitrogen nucleophiles to form 1,1-diamino derivatives (Scheme 47).
When Ar1 (Scheme 47) is an electron-withdrawing system, such as 4-nitrophenyl, 2- pyridyl, 2-thiazolyl and 2-benzothiazolyl, aminals (157) are obtained in high yields from the reaction mixture of aldehydes and heteroaryl amines in methanol or in DMSO (without

414 |
|
Luciano Forlani |
|
|
|
|
|
|
|
|
|
Ar |
H |
|
Ar |
Ar1 |
|
|
|
NHAr1 |
|||
C O + Ar1 |
N |
− H2 O |
|
+ A r1NH2 |
|
|
NHAr1 |
||||
|
C N |
|
|
Ar |
|
C |
|
||||
|
|
|
|||||||||
|
|
|
|
|
|||||||
H |
H |
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
||||||
|
|
|
|
|
|
|
|
||||
|
|
(156) |
|
(157) |
|
||||||
SCHEME 47
catalysts)220. Probably the electron-withdrawing moiety of imines (156) exerts an activating effect on the CDN bond, as shown in 158. This appears more prone to nucleophilic attack than the CDO bond, even when electrophilic catalysis is not operating.
N |
H |
N |
|
H |
|
|
− |
+ |
|||
N |
C |
N |
C |
||
|
|||||
S |
Ar |
S |
|
Ar |
(158)
This explanation confirms the usual findings that primary amines give Schiff bases without further complications, while secondary amines (piperidine) and some aromatic aldehydes easily form the gem-diamino derivatives214 (160), probably through the immonium cation (159) that is usually accepted in enamine formation (equation 31).
H
C N +
Ar
(159)
H
Pip erid in e
Ar C N
N
(31)
(160)
When the reaction products of the reaction involve the nitrogen atoms of nucleophiles in the formation of heterocyclic derivatives, the formation of 1,1-diamino derivatives is usually used to prepare polyazacyclic derivatives221. Scheme 48 shows222 the formation of fused polyazapolycyclic compounds through a condensation between 1,3-diaminopropane and glyoxal to prepare the 1,1-diamino derivative 161. The subsequent reaction of 161 with formaldehyde forms fused addition products (162).
Some authors suggest that the addition of alcohols with amines223 to some imines containing heterocycles occurs without catalysis.
Another instance of a stable bis-adduct of the carbonyl group of aldehydes is the condensation between aromatic aldehydes and electron-rich thiazole derivatives224 (Scheme 49) where the nucleophilic centre is carbon 5 of the thiazole ring. When carbon 5 of the thiazole ring is activated by an electron-donating amino or hydroxy group (as in 163) and in the presence of small amounts of HCl, to activate the carbonyl group, compounds (164) are obtained by non-reversible condensation. The reaction of the amino group (bonded in
|
8. Complex formation involving double-bonded functional groups |
415 |
|||||||||||||||
|
|
|
|
|
|
H |
O |
|
|
|
|
|
|||||
CH2 |
|
NH2 |
|
|
C |
|
|
|
|
|
HN |
NH |
|
|
HN |
N |
|
|
|
|
|
|
|
|
|
|
|
+HCHO |
|
|
|||||
CH2 |
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
C |
|
|
|
|
|
|
|
|
|
|
|
|||
CH2 |
|
NH2 |
|
|
|
|
|
|
|
HN |
NH |
|
|
|
|
||
|
|
|
H |
|
|
|
|
|
|
|
HN |
N |
|||||
|
|
|
|
|
|
O |
|
|
|
|
|
||||||
|
|
|
|
|
|
|
(161) |
|
|
(162) |
|||||||
|
|
|
|
|
|
|
|
|
|
SCHEME 48 |
|
|
|
|
|
||
|
|
|
|
|
|
N |
|
|
|
H+ |
N |
H |
N |
|
|||
ArCHO + |
|
|
|
X |
C |
|
|||||||||||
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
S |
|
|
|
|
|
X |
S |
Ar |
S |
X |
|
|
|
|
|
(163) |
|
|
|
|
|
|
|
(164) |
|
|
|
||
ArCHO = benzaldehyde, 4-methylbenzaldehyde, 4-methoxybenzaldehyde |
|
|
|||||||||||||||
X = |
|
|
NH2 , |
|
|
NMe2 , |
|
OH |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
||||||||
SCHEME 49
position 2 of the thiazole ring) on the carbonyl group is illustrated by Scheme 47 and is clearly reversible.
A recent instance225 of reaction 22 (X D NH) involves the reactions of some (heteroarylchloromethyl)lithium (165) reagents with imines (166) to form 167 and to produce the heteroaryl aziridines (168) as depicted in Scheme 50. Aziridines (167) are obtained in the preferential (or exclusive) conformation E. A tentative explanation of this behaviour is the different steric compression in the transition states affording isomeric E or Z aziridines.
|
|
|
|
|
|
H |
|
Cl |
|
|
Ar |
|
R2 |
|
|
H |
|
|
|
|
1 |
|
|
||||
|
|
|
R1 |
|
|
Ar |
C |
C |
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
||
Ar |
|
C Li + |
C NR |
|
Li+ |
− |
2 |
|
H |
N |
R1 |
||
|
|
|
|||||||||||
|
|
|
|
|
|
||||||||
|
|
|
R2 |
|
|
N |
|
R |
|
|
|
|
|
|
|
Cl |
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(165) |
|
(166) |
|
|
(167) |
|
|
|
|
(168) |
|
|
Ar = 2-pyridyl, 2-quinolyl, 2-benzothiazolyl
R = Ph
R1, R2 = cyclopentylidene, cyclohexylidene, fluorenylidene
R1 = H, R2 = Ph
R1 = R2 = Me
R1 = Ph, R2 = Me
SCHEME 50

416 |
Luciano Forlani |
VII. REFERENCES
1.J. E. Leffler and E. Grunwald, Rates and Equilibria of Organic Reactions, Wiley, New York, 1963.
2.C. Reichardt, Solvent Effects in Organic Chemistry, Verlag Chemie, New York, 1979.
3.V. Gutmann, Coordination Chemistry in Non-Aqueous Solvents, Springer, Wien, 1968.
4.V. Gutmann. The Donor Acceptor Approach to Molecular Interaction, Plenum Press, New York, 1978.
5.C. Reichardt, Chem. Rev., 94, 2319 (1994).
6.M. Patz, H. Mayr, J. Maruta and S. Fukuzumi, Angew. Chem., Int. Ed. Engl., 34, 1225 (1995).
7.J. K. Kochi, Angew. Chem., Int. Ed. Engl., 27, 1227 (1988).
8.G. A. Jeffrey and W. Soenger, Hydrogen Bonding in Biological Molecules, Springer-Verlag, Berlin, 1991.
9.H. K. Hall and A. B. Padias, Aldrichim. Acta, 28, 37 (1995).
10.A. D. Attie and R. T. Raines, J. Chem. Educ., 72, 119 (1995).
11.S. L. Schreiber, Chem. Eng. News, 22 October 26 (1992).
12.W. P. Jencks, Acc. Chem. Res., 13, 161 (1980).
13.L. Pauling, The Nature of the Chemical Bond, Cornell Press, New York, 1959.
14.G. C. Pimentel and A. L. McClellan, The Hydrogen Bond, W. H. Freeman Co., San Francisco, 1960.
15.R. F. Foster, Organic Charge Transfer Complexes, Academic Press, New York, 1969.
16.P. A. Kollman, Acc. Chem. Res., 10, 365 (1977).
17.P. Hobza and R. Zahradn´ık, Chem. Rev., 88, 871 (1988).
18.A. D. Buckingham and P. W. Fowler, Can. J. Chem., 63, 2018 (1985).
19.P. Hobza and R. Zahradn´ık, Intermolecular Complexes, Chap. I, Elsevier, Amsterdam, 1988.
20.D. J. Sandman and A. F. Richter, J. Am. Chem. Soc., 101, 7079 (1979).
21.N. Heinrich, W. Koch, G. Frenking and H. Schwarz, J. Am. Chem. Soc., 108, 593 (1986).
22.K. N. Houk, Acc. Chem. Res., 8, 361 (1975).
23.A. B. Padias and H. K. Hall, J. Org. Chem., 52, 4536 (1987).
24.D. Philp, V. Gramlich, P. Seiler and F. Diederich, J. Chem. Soc., Perkin Trans. 2, 875 (1995).
25.G. A. Derwish, R. S. Salman and M. S. Al-Marsoumi, J. Inclusion Phenom. Mol. Recognit.
Chem., 20, 123 (1994).
26. J. F. Frey, A. M. Andrews, D. G. Ankoviac, D. N. Beaman, L. E. Du Pont, T. E. Elsner, S. R. Lang, M. A. Oosterbaan Zwart, R. E. Seagle and L. A. Torreano, J. Org. Chem., 55, 606 (1990).
27.E. F. Hilinski, J. M. Masnovi, C. Amatore, J. K. Kochi and P. M. Rentzepis, J. Am. Chem. Soc., 105, 6167 (1983).
28.E. M. Kosower, Prog. Phys. Org. Chem., 3, 81 (1965).
29.T. Kim, H. Sarker and N. L. Bauld, J. Chem. Soc., Perkin Trans. 2, 577 (1995).
30.N. Mataga, Biomolecules, Japan Sci. Soc. Press, Tokyo; Elsevier, Amsterdam, 1985, p. 127.
31.J. D. Simon and K. S. Peters, Acc. Chem. Res., 17, 277 (1984).
32.B. K. Adams and W. R. Cherry, J. Am. Chem. Soc., 103, 6904 (1981).
33.J. L. Goodman and K. S. Peters, J. Am. Chem. Soc., 107, 1441 (1985).
34.H. Hayashi and S. Nagakura, Chem. Phys. Lett., 53, 201 (1978).
35.W. Hub, S. Schneider, F. Dorr,¨ J. D. Oxman and F. D. Lewis, J. Am. Chem. Soc., 106, 701 (1984).
36.W. Hub, S. Schneider, F. Dorr,¨ J. D. Oxman and F. D. Lewis, J. Am. Chem. Soc., 106, 708 (1984).
37.L. L. Rodina and A. V. Ryzhakov, Heterocycles, 40, 1035 (1995).
38.L. Mathew, B. Varghese and S. Sankararaman, J. Chem. Soc., Perkin Trans. 2, 2399 (1993).
39.N. Kornblum, P. Ackermann and R. T. Swiger, J. Org. Chem., 45, 5294 (1980)
40.L. Eberson, M. P. Hartshorn, F. Radner and W. T. Robinson, J. Chem. Soc., Chem. Commun., 566 (1992).
41.L. Eberson and F. Radner, J. Am. Chem. Soc., 113, 5825 (1991).
42.C. P. Butts, L. Eberson, M. P. Hartshorn, O. Persson and W. T. Robinson, Acta Chem. Scand., 49, 253 (1995).
43.A. Guy, M. Lemaire and J-P. Guette,´ Tetrahedron, 38, 2339 (1982).
44.A. Guy, M. Lemaire and J-P. Guette,´ Tetrahedron, 38, 2347 (1982).