

23. The thiocarbonyl group |
1425 |
8. C,halogen cleavage
Here we report the thionation of amidinium chlorides (Vilsmeier salts) with sodium sulfide to give the corresponding enaminothioketones349,350 as shown in equation 73349.
Cl S
+ |
Na2 S |
|
|
|
|
|
|
(73) |
||||||
|
|
|||||||||||||
Ar C CH CH NR2 |
|
Ar |
|
C |
|
CH |
|
CH |
|
NR2 |
||||
|
|
|
|
|||||||||||
|
|
|
|
|
||||||||||
Cl− |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A new method for the preparation of diethyl thioxomalonate, already prepared from Bunte salts340 (Section III.E.5), has been introduced by Abelman351. In this approach, diethyl chloromalonate was reacted with cesium carbonate in the presence of elemental sulfur to yield the thioxoester which underwent a facile [4C2] cycloaddition (equation 74).
EtO2 C CO2 Et Cs2 CO3 |
EtO2 C |
CO Et |
−CsCl |
EtO C |
CO Et |
|
|
|
2 |
2 |
2 |
||
|
S8 |
Cl |
S− Cs+ |
|
|
S |
|
|
|
||||
Cl |
|
|
|
|
||
(74)
F. Cycloreversion Reactions
As stated by Schaumann1, cycloreversion is the thermally or photochemically induced cleavage of two bonds in a carboor heterocyclic ring without involvement, in general, of a reagent. Fragments with bonds are formed and, under the appropriate conditions, this strategy, together with the direct thionation of carbonyl compounds, represents the most common method for the preparation of a wide array of thiocarbonyl compounds.
1. [2+1] Cycloreversion
Hypervalent three-membered thiaheterocycles have been claimed as intermediates which undergo cheletropic extrusion of thiones352. This approach, however, has found scant application as a synthetic method.
2. [4+1] Cycloreversion
This route has been commonly used for the preparation of ˛,ˇ-unsaturated thioketones such as o-thioquinonemethides353 69, which were trapped with dienophiles as illustrated in equation 75.
3. [2+2] Cycloreversion
[2C2] Cycloreversions are well documented in the organic literature and were reviewed by Schaumann and Ketcham354 a number of years ago. This strategy has been primarily applied to the synthesis of thioquinone derivatives and thioketenes. Several precursors to these compounds have been reported. Thus, 1,2-oxathietanes 70, which are isolable at moderate temperatures in organic solvents, undergo a formal [2 C 2] cycloreversion via a biradical species to yield monothio-o-benzoquinones355,356 71 (equation 76). This process is a valence tautomerization which occurs spontaneously under the experimental conditions employed.

1426 M. T. Molina, M. Ya´nez,˜ O. Mo,´ R. Notario and J.-L. M. Abboud
|
|
O |
O |
|
|
N |
|
hν |
|
Ph |
|
O |
|
|
|
S |
|
S |
|
|
(69) |
|
(75) |
|
O |
|
|
|
|
|
|
|
|
N |
Ph |
|
|
S |
|
|
|
O |
|
S |
|
S |
|
|
|
|
|
O |
|
O |
(76) |
|
|
|
|
(70) |
|
(71) |
|
Similarly, thietanes have been the subject of intense studies aiming at the preparation of o-thiobenzoquinone methides 69, which can be prepared by thermal ring opening of benzothietes 72 as depicted in equation 77357 359. The dienes 69 thus formed can be trapped by dienophiles in hetero-Diels-Alder reactions or undergo spontaneous dimerization. Very recently, thermally generated 69 was applied to the preparation of fullerene derivatives360. Also, the parent compound, thietane 73, and the corresponding deuterated derivative have been the subject of photochemical studies361. Photolysis of thietane in an argon-matrix at low temperature proved to be a clean source of thioformaldehyde which usually polymerizes to a cyclic trimer (trithiane) under normal conditions (equation 78).
|
|
|
S |
|
S |
|
S |
∆ |
|
dimerize |
|
|
|
|
|
||
|
|
hν |
|
|
|
|
|
|
CH2 |
|
S |
(72) |
|
(69) |
|
|
(77) |
|
|
|
|
|
|
|
|
S |
hν |
H |
|
|
|
|
S |
(78) |
|
|
|
|
12 K |
||
|
|
|
H |
||
|
|
|
|
||
|
|
|
|
|
|
|
|
(73) |
|
|
|
Quantum chemical studies of these electrocyclic ring-opening355,356,362 show that benzothietes are thermodynamically more stable than o-thioquinonemethides.
1,2-Dithietanes 74 have been studied as appropriate sources for their valence tautomers 1,2-dithiones 75a, 75b (equation 79) and theoretical calculations have been carried
23. The thiocarbonyl group |
1427 |
out363,364. Further details are given in Section II.
H |
|
H |
S |
H |
S |
|
S |
||||
|
|
|
|
|
|
|
S |
H |
S |
S |
(79) |
H |
|
H |
|||
|
|
|
|
|
|
|
(74) |
|
(75a) |
|
(75b) |
Studies in the benzene series have been reported by Bock and Rittmeyer365 by carrying out the thermal fragmentations in the gas phase. Finally, few examples of the application of [2C 2] cycloreversions in the preparation of thioketenes have been reported321. Irradiation in an argon-matrix of 2,3-thiophenedicarboxylic acid anhydride afforded, among other products, thioketene (76) in 20% yield.
• |
• |
• |
S |
(76)
4. [2+3] Cycloreversion
Five-membered heterocycles have been frequently used in the generation of thiocarbonyl compounds, and 1,3-dithiolanes and 1,3-dithianes are the most common precursors for these transformations which have been reviewed by Schaumann366. These [3 C 2] cycloreversions are based on the generation of a negative charge, by using a strong base (BuLi is preferred), on the five-membered ring. The resulting anion gives an instantaneous cleavage into a thioketone along with an anionic fragment 77 (equation 80).
R1 |
S |
|
BuLi |
R1 |
S |
|
||
|
|
|
|
|
|
|
||
R2 |
S |
|
|
|
R2 |
S |
− |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
(80) |
|
|
R1 |
|
|
|
|
||
|
|
|
S + −S |
|
CH |
|
CH2 |
|
|
|
|
|
|||||
|
|
|
|
|||||
|
|
|
|
|||||
R2
(77)
Since ketones are readily converted into 1,3-dithiolanes by reaction with 1,2- ethanedithiol and BF3.Et2O as catalyst, this is a flexible method for the preparation of thioketones. However, this type of chemistry cannot be applied to the synthesis of thioaldehydes because treatment with base of a dithiolane (or derivative) formed from an aldehyde (R1 or R2 D H) preferentially gives an ‘umpolung’367 reaction by removal of the acidic hydrogen ˛ to both sulfur atoms223. In the case of thioaldehydes, 4,5- disubstituted dithiolanes with electron-withdrawing groups have been utilized in order to avoid ‘umpolung’ reactions.
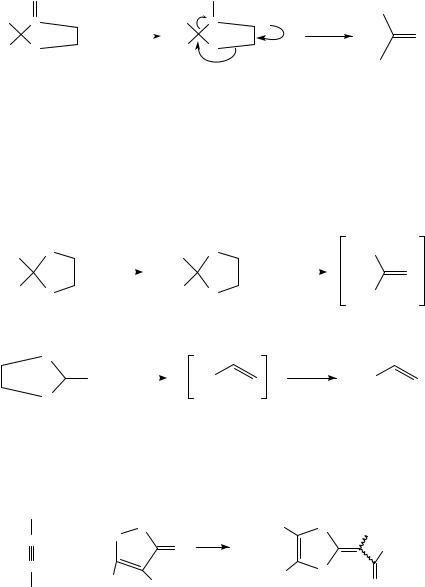
1428 M. T. Molina, M. Ya´nez,˜ O. Mo,´ R. Notario and J.-L. M. Abboud
Milder conditions for this cleavage have been achieved by using more reactive analogs such as sulfonium salts or silyl derivatives of the corresponding sulfoxides. The latter strategy is depicted in equation 81368.
|
O |
|
|
OTBDMS |
|
|
R1 |
S |
|
R1 |
S |
R1 |
|
TBDMSTfO |
− |
|||||
|
|
|
+ |
|||
|
|
|
|
|
S |
|
2 |
S |
EtNPri2 |
2 |
S |
||
|
||||||
R |
|
|
R |
|
R2 |
|
|
|
|
|
|
(81)
The corresponding 1,3-dithiolane S-oxides were prepared by oxidation of the thioacetals with m-chloroperbenzoic acid (mCPBA) and, notably, this approach has proven successful in the obtention of both thioaldehydes and thioketones.
Pyrolytic methods involve the use of dithiolane S, S-dioxides as starting materials which, upon heating, afford reactive thioaldehydes, such as trifluorothioacetaldehyde, trapped as the Diels-Alder adduct369,370 (equation 82). In equation 80 an enethiolate was formed, and this feature has been employed also in the cleavage of the oxathiolane 78. The silver vinylthiolate 79 thus obtained was applied in the preparation of new antibacterial cephem derivatives371.
|
|
|
|
O2 |
|
F3 C |
F3 C |
S |
F3 C |
S |
|
||
|
KMnO4 |
|
750˚C |
|||
|
|
|
|
|||
|
|
|
|
|
|
S |
|
|
|
|
|
|
|
H |
S |
H |
S |
−SO2 , −C2 H4 |
||
|
H |
|||||
|
|
|
|
|
(82) |
|
|
S |
|
|
|
|
|
|
LDA |
||
OEt |
|
|
LiS |
−78 |
|
||
|
°C |
||
O
A gNO3
Ag S
(78) |
(79) |
A related reaction is the cycloaddition of 1,2-dithiole-3-ones 80 onto a number of electrophilic alkynes such as acetylenedicarbaldehyde or its monoacetal. It affords different stable thials or thiones372 (equation 83).
CH(OEt)2 |
|
S |
OHC |
S |
|
|
|
R1 |
|||
C |
|
S |
|
|
R2 |
+ |
|
S |
|
||
|
|
|
|
||
C |
R2 |
|
(OEt)2 CH |
S |
(83) |
CHO |
R1 |
|
S |
||
|
|
|
|
|
R1 , R2 = H, Me, Ar
(80)
23. The thiocarbonyl group |
1429 |
5. [2+4] cycloreversion. Flash Vacuum Thermolysis (FVT)
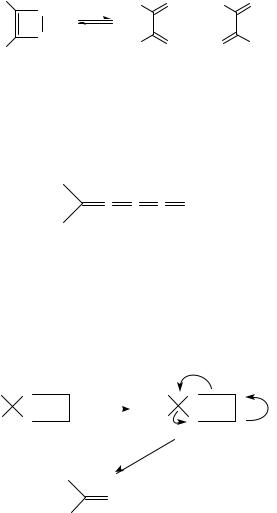
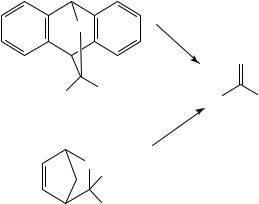
Retro-Diels-Alder reactions giving thiocarbonyl compounds are favored when simultaneously a comparatively stable diene is formed1. This is the case with anthracene and cyclopentadiene Diels-Alder adducts 81 and 82 which, upon heating, afford a wide array of thioaldehydes and thioketones (equation 84). These adducts are stable at room temperature and have become a convenient way of storing very reactive thiocarbonyl compounds. Cyclopentadiene is the cheapest and most reactive diene for use in Diels-Alder reactions. Also, strain in the bridged cycloadducts facilitates retro-Diels-Alder cleavage224.
S |
∆ |
|
|
|
|
|
|
S |
R |
R′ |
R′ |
R |
||
(81) |
∆ |
(84) |
|
S R
R′
(82)
Besides common pyrolytic techniques to decompose these cycloadducts, in recent years Flash Vacuum Thermolysis (or Pyrolysis) (FVT or FVP) has found increasing application20,373. FVT is a very clean and efficient way of generating (in the gas phase) transient species for spectroscopic or chemical investigations. It generally involves using temperatures in excess of 400 °C and pressures of less than 10 4 mbar for the vacuum pyrolysis, and isolation of the free thiocarbonyl compound in a liquid nitrogen trap or argon matrix223. In practice, a precursor is vaporized under vacuum in an oven where it is cleaved into (generally) two molecules. One of them is the molecule to be studied which can be trapped at the oven exit by an appropriate volatile reagent (such as another diene) or spectroscopically characterized either in the gas phase (IR, PES, MS,
. . .) or in the condensed phase after cooling, as mentioned above (IR, UV, NMR). The second molecule formed must be unreactive towards the one being studied and, ideally, they should be easily separable, for instance by selective crystallization of the unreactive by-product. However, simple volatile by-products whose spectra are well known and do not hinder the desired identification, such as N2 or ethylene, are acceptable20.
Thermal cycloreversion of the adducts can be accomplished at a convenient rate when heated in toluene under reflux. If a new diene is present in the reaction mixture, the thioaldehyde thus generated in the retro-Diels-Alder reaction may give a new adduct. Therefore, adducts 81 and 82 act as thioaldehyde or thioketone transfer reagents. These adducts dissociate reversibly on heating, thus ensuring that the concentration of the labile species remains very low. For this reason, polymerization is not a serious problem especially in the case of thioaldehydes224. The transient thiocarbonyl compounds can be trapped not only by dienes but also by 1,3-dipolar cycloadditions332 (equation 85).
1430 M. T. Molina, M. Ya´nez,˜ O. Mo,´ R. Notario and J.-L. M. Abboud
+
N
BocNH |
+ |
N
−
S
O
RO2 C |
H |
(85) |
toluene |
|
|
∆ |
|
|
|
N |
BocNN |
S |
|
N |
O |
CO2 R |
R = H, Et, allyl |
|
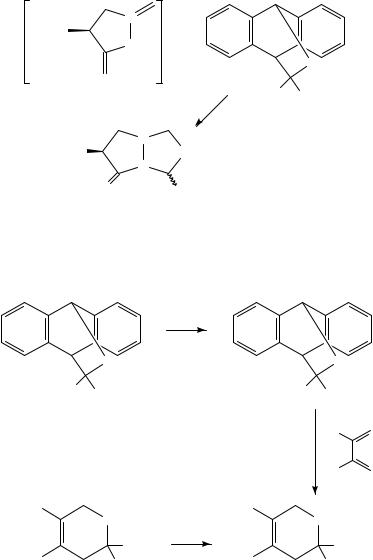
An interesting feature of adducts 81 and 82 is that they can be additionally functionalized by alkylation with a strong base, usually LDA, using a range of alkyl halides (equation 86)224,374, thus providing a route for the generation of reactive thioketones. Kirby and coworkers have applied the thermal cycloreversion in the synthesis of thiashikimic acid derivatives375 (equation 87).
|
|
1. LDA |
|
|
|
2. RBr |
|
|
S |
|
S |
H |
CO2 Et |
R |
CO2 Et |
|
|
(83)
Me
S
1. LDA
CO2 Et
2. RBr
Me H
R = Me, Et, allyl, benzyl
Me
111 ˚C |
(86) |
|
|
|
Me |
Me
S
CO2 Et
Me R
Unsaturated 1,3-dithianes also afford thioketones on heating215.
FVT, initially applied to the synthesis of methanethial and the corresponding S-oxide376, was also the key reaction for the generation of thioxoethanal377 and thioxoethanal S-oxide378 using as precursors the corresponding 2,3-dihydro-1,4-oxathiin derivatives (equation 88). The reaction products were characterized by chemical trapping with dienes and by PE spectroscopy in addition to low-temperature IR and NMR spectroscopy.
23. The thiocarbonyl group
|
|
|
S |
|
83 |
111 ˚C |
|
|
|
|
|
|
||
−anthracene |
H |
CO2 Et |
||
|
A cO OA c
OA c
CO2 Et
|
|
AcO |
|
|
S |
|
|
OAc |
S |
H |
S |
FVT |
|
S |
720 ˚C |
|
|
−C2 H4 |
|
H |
O |
|
|
H |
O |
CHO
53%
1431
(87)
(88)
The same strategy has also been applied to the preparation of ˛-oxothiones379 and thioformyl cyanide89 (equation 89), which were characterized by PE and IR spectroscopy.
|
FVT |
S |
|
S |
(89) |
||
|
|||
|
H |
CN |
|
CN |
− |
|
|
|
|
Thioketenes were generated by FVT of the bridged cycloadducts and were trapped with dimethylamine288 as shown in equation 90. Thiochromane 84 underwent complete cycloreversion under FVT conditions affording o-thioquinonemethide 69, which spontaneously dimerized380 (equation 91).
FVT |
|
|
|
|
|
|
|
S |
700 ˚C |
|
|
|
|
|
HNMe2 |
|
|
H2 C |
|
C |
|
S |
H3 C C |
(90) |
||
|
|
|
||||||
|
|
|
|
|||||
|
|
|
|
|
NMe2
S −
1432 M. T. Molina, M. Ya´nez,˜ O. Mo,´ R. Notario and J.-L. M. Abboud
S S
FVT, 1000 °C
−C2 H4
CH2
(84) |
(69) |
(91)
S
S
G. Reductive C,S Cleavage
Aqueous sodium sulfide reduced the aryl thiocyanate 85, prepared from ethidium bromide, affording the air-sensitive dithiol 86 in 63% yield381 (equation 92). Also, Nicolaou’s group has reported on the cleavage of dithiatopazine 87, a stable 1,2-dithietane system with loss of one sulfur atom and formation of a rearranged thioketone 88382 (equation 93).
NCS |
|
|
SCN |
HS |
|
S |
|
|
|
|
Na2 S |
|
|
|
|
N + |
|
|
N |
|
Ph |
|
Et |
|
Ph |
|
Et |
|
(85) |
|
|
|
(86) |
(92) |
|
|
|
H |
|
|
O |
|
|
|
|
|
|
|
|
|
|
O |
|
H |
H |
H |
|
S |
|
|
||
O |
|
PPh3 |
|
|
||
|
|
|
O |
|
||
|
|
S |
|
H |
|
|
|
|
|
O |
|
||
|
|
|
|
|
||
|
|
O |
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
S |
H |
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
H |
|
|
|
(87) |
|
|
(88) |
(93) |
|
|
|
|
|
|
|
|
H. Sigmatropic Shifts
1. [1,2] Shifts
Thioketenes can be formed by 1,2-sigmatropic shifts occurring through the four-electron species 89 which may be considered as a diradical, as a 1,3-dipole or as a carbene1. In
23. The thiocarbonyl group |
1433 |
addition, 89 may cyclize to a thiirene 90 (equation 94).
R1 |
R1 |
|
|
R1 |
|
|
4e− |
|
|
S |
|
~R2 |
||
|
|
|
C C S |
|
|
|
|
|
(94) |
2 |
R2 |
S |
|
R2 |
R |
|
|
|
|
(90) |
|
(89) |
|
|
The most convenient sources of species 89 are 1,2,3-thiadiazoles 91, which are valence tautomers of the unknown ˛-diazothioketones. Loss of nitrogen from 91 may be achieved by irradiation or by thermolysis. Larsen and coworkers383 examined the irradiation of 91 in the presence of diethylamine and isolated N, N-diethylthioacetamide in high yield, which implies trapping of thioketenes during photolysis. Mechanistic studies excluded thiirene as the intermediate in the photolysis at 150 K (equation 95).
N |
|
H |
|
Et2 NH |
|
S |
|||||
|
hν |
|
|
|
|
|
|
|
|||
|
|
C C |
S |
|
|
|
|
|
|
|
|
N |
|
|
CH3 |
|
C |
|
NEt2 (95) |
||||
|
H |
|
|
|
|
||||||
S |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(91) |
|
|
|
|
|
|
|
|
|
|
|
2. [1,3] Shifts
Thioketenes may be formed from 1-alkynyl disulfides by [1,3]sigmatropic shift at78 °C384 (equation 96), and are generally stable in solution at room temperature for a few hours.
|
|
|
|
|
|
|
|
|
|
|
R1 |
(96) |
R1 |
|
|
|
|
S |
|
S |
|
R2 |
|
C C S |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
||||||
|
|
|
R2 S
3. [2,3] Shifts
Block and Zhao reported the thermal transformation of diallyl sulfoxide (92) into 3,4-dihydro-2H-thiopyrans, the thioacrolein Diels-Alder adducts, in good to excellent yields385 (equation 97).
O− |
|
O |
|
|
S+ |
|
|
||
S |
|
|
||
|
90−110 °C |
|
N Ph |
|
|
|
|
(97) |
|
|
NPM |
|
||
|
|
|
||
|
|
91% |
O |
|
|
|
|
|
|
(92) |
|
|
(93a) |
|
1434 M. T. Molina, M. Ya´nez,˜ O. Mo,´ R. Notario and J.-L. M. Abboud
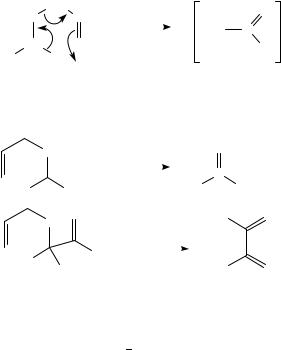
The reaction does not afford the expected product, thioacrolein S-oxide, but the thial, and the authors propose as mechanism the initial and reversible [2,3]sigmatropic rearrangement of the allylic sulfoxide to give an allyl sulfenate which undergoes attack by a second molecule of sulfoxide. Further transformations, such as trapping with N-phenylmaleimide (NPM), of this intermediate yield 93a. This result is in striking contrast with the pyrolytic behavior of allylvinyl sulfone which undergoes [3,3]sigmatropic rearrangement386, and diallyl sulfide which suffers a retro-ene reaction387.
I. Retro-ene Reactions
In recent years some applications of retro-ene reactions in the generation of thiocarbonyls have appeared. The retro-ene reaction is a symmetry-allowed process which resembles both [2 C 4] cycloreversion and a [1,5] sigmatropic shift of hydrogen. It usually requires high temperatures, therefore FVT has become very widespread in these reactions.
Flash pyrolysis in an argon matrix of allylethylsulfide afforded thioacetaldehyde characterized by comparison of experimental and calculated IR spectra182 (equation 98) (see Section II).
|
CH2 |
|
|
|
S |
|
S |
CH |
800 |
°C |
|
||
CH3 C |
(98) |
|||||
|
|
|||||
HC |
CH2 |
−propene |
||||
|
|
|||||
|
|
|
H |
|||
H3 C |
H |
|
|
|
||
|
|
|
|
|||
Thioformyl cyanide was generated under FVT conditions by retro-ene cleavage of allyl cyanomethyl sulfide as shown in equation 99. Spectroscopic properties of this compound are discussed in Section II. Note that thioformyl cyanide had already been prepared by retro-Diels-Alder reaction (see Section III.F.5).
S S
FVT, 850 °C
|
|
|
|
|
|
C |
(99) |
|
|
−propene |
|||||
|
|
|
|||||
H |
CN |
|
|
|
H |
|
CN |
|
|
|
|
|
|
||
|
O |
|
|
|
|
CH3 |
S |
|
S |
|
|
|
|
|
|
|
|
|
FVT, 800 °C |
|
|
||
|
R |
|
|
|
|
(100) |
|
H |
|
−propene |
|
||||
CH3 |
|
|
|
|
R |
O |
|
|
|
|
|
|
|||
R = H, Me
In similar way, the simplest ˛-oxothione was obtained by retro-ene reaction of the corresponding allyl sulfide379 (equation 100) and characterized by IR spectroscopy. Upon FVT at higher temperatures (R D Me, 900 1000 °C) subsequent loss of carbon monoxide took place yielding thioacetone379.
J. Isomerizations
An original isomerization of thiadiazolidine-ones into triazolidin-one-thiones has been achieved enzymatically388. The enzymes used were bovine and equine glutathione