

2009.-Byelorussian Pharmacopoeia_Volume 3
.pdfТроксерутин |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
591 |
|||||
|
|
|
|
|
|
|
H |
NH2 |
|
|
K. R = H: (S)-2-Амино-3-[2-(1H-индол-3-илме- |
|||||||||||||||
|
|
|
H |
|
|
|
||||||||||||||||||||
|
|
|
N |
CO2H |
тил)-1H-индол-3-ил]пропановая кислота. |
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
F. (S)-2-Амино-3-(фениламино)пропановая |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NH |
|||||||||
кислота (3-фениламиноаланин). |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
HN |
|
OH |
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|||||
|
|
|
|
|
H |
NH2 |
|
|
|
|
|
|
|
|
|
|
N |
|
NH |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
CO2H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CO2H |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
G.(S)-2-Амино-3-(2-гидрокси-1H-индол-3-ил)про- |
|
|
L. 1-(1H-Индол-3-илметил)-1,2,3,4-тетрагидро- |
|||||||||||||||||||||||
пановая кислота (2-гидрокситриптофан) |
9H-β-карболин-3-карбоновая кислота. |
|||||||||||||||||||||||||
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
NH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
ТРОКСЕРУТИН |
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
CO2H |
Troxerutinum |
|
|
|
|
|
|
|||||||||||
H. R = H: (3RS)-1,2,3,4-Тетрагидро-9H-β-кор- |
|
|
|
|
|
|
||||||||||||||||||||
TROXERUTIN |
|
|
|
|
|
|
||||||||||||||||||||
болин-3-карбоновая кислота. |
|
|
|
|
|
|
||||||||||||||||||||
I. R = CH3: 1-Метил-1,2,3,4-тетрагидро-9H-β- |
|
|
|
|
|
|
|
|
|
|
|
|
HO |
|
|
|
O |
|||||||||
карболин-3-карбоновая кислота. |
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
OH |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
OH |
|
O |
|
|
|
|
|
|
||||||||||||||
|
HN |
|
|
|
|
|
|
O |
|
|
|
|
|
|
O |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
NH |
|
CH3 |
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
OH |
|
OH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
OH |
|
|
OH |
|
|
|
|
|
OH |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
CO2H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
H2N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
J. R = CHOH-CH2-OH: (S)-2-Амино-3-[2-[2,3- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
||||||||||||
дигидрокси-1-(1H-индол-3-ил)пропил]-1H-индол- |
|
|
|
|
|
|
|
|
HO |
|
|
|
|
|
|
|||||||||||
3-ил]пропановая кислота. |
|
С33Н42О19 |
|
|
|
|
|
М.м. 743 |
||||||||||||||||||
Пропускание
94
92
90
88
86
84
82
80
78
76
74
72
70
68
66
64
62
60
58
56
54
3000 |
2000 |
1500 |
500 |
Волновое число (см-1)
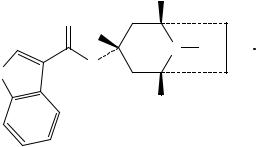
Рисунок 1. Инфракрасный спектр пропускания ФСО троксерутина.
592 |
Государственная фармакопея Республики Беларусь |
ОПРЕДЕЛЕНИЕ
Троксерутин представляет собой смесь
О-гидроксиэтилированных производных рутина,
содержащую не менее 80,0% 2-[3,4-бис(2- гидроксиэтокси)фенил]-3-[[6-О-(6-дезокси- α-L-маннопиранозил)-β-D-глюкопиранозил]- окси]-5-гидрокси-7-(2-гидроксиэтокси)-4Н-1-
бензопиран-4-он(трис(гидроксиэтил)рутин), и
содержит не менее 95,0% и не более 105,0% С33Н42О19 в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Желтовато-зеленый кристаллический порошок. Гигроскопичен.
Легкорастворим в воде, малорастворим в
96% спирте, практически нерастворим в мети-
ленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Абсорбционная спектрофотометрия в инфракрасной области (2.2.24).
Сравнение: ФСО троксерутина # или
спектр, представленный на рисунке 1.
В. Просматривают хроматограммы, полученные в испытании «Состав». Основной пик на хроматограмме испытуемого раствора
по времени удерживания и площади соот-
ветствует основному пику на хроматограмме раствора сравнения (а).
ИСПЫТАНИЯ
Состав. Жидкостная хроматография (2.2.29): определение проводят методом внутренней нормализации.
Испытуемый раствор. 10,0 мг испытуемого образца растворяют в подвижной фазе,
при необходимости используя ультразвуко-
вую баню, и доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (а). 10,0 мг ФСО троксерутина растворяют в подвижной фазе, при необходимости используя ультра-
звуковую баню, и доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (b). 1 мл раствора
сравнения (а) доводят подвижной фазой до
объема 10 мл. 1 мл полученного раствора доводят подвижной фазой до объема 100 мл.
Условия хроматографирования:
–колонка длиной 0,25 м и внутренним диаметром 4,6 мм, заполненная силикагелем октадецилсилильным эндкепированным для хроматографии Р, с размером частиц 5 мкм;
–подвижная фаза: смесь из 20 объемов ацетонитрила Р и 80 объемов раствора 15,6 г/л натрия дигидрофосфата Р, доведенного кислотой фосфорной разведенной Р или раствором натрия гидроксида разведенным Р до рН 4,4;
–скорость подвижной фазы: 0,5 мл/мин;
–спектрофотометрический детектор, длина волны 350 нм;
–объем вводимой пробы: 10 мкл;
–время хроматографирования: 2-крат-
ное время удерживания основного компонента троксерутина (трис(гидроксиэтил)рутин).
Относительное удерживание (по отношению к трис(гидроксиэтил)рутину; время удерживания — около 25 мин): тетракис-
(гидроксиэтил)-рутин |
— |
около |
0,5; |
моно(гидроксиэтил)рутин |
— |
около |
0,8; |
бис(гидроксиэтил)рутин — около 1,1. |
|
||
Пригодность хроматографической |
си- |
||
стемы: раствор сравнения (а): |
|
|
|
–коэффициент разделения пиков: не ме-
нее 2,0 (Hp — высота пика бис(гидроксиэтил)- рутин относительно базовой линии; Hv — рас-
стояние между базовой линией и нижней точкой
кривой, разделяющей пики бис(гидроксиэтил)
рутина и трис(гидроксиэтил)рутина).
–отношение сигнал/шум: не менее 10
для основного пика на хроматограмме раствора сравнения (b).
Состав:
–основной пик: не менее 80 %;
–любые другие пики: не более 5 %; допускается присутствие одного пика более 5 %, но менее 10 %.
На хроматограмме испытуемого раствора
не учитывают пики с площадью менее площади основного пика на хроматограмме раствора сравнения (b).
Этиленоксид. Не более 0,0001 %
(1 ррm). Парофазная газовая хроматография (2.2.28).
Испытуемый раствор. К 1,00 г испытуемого образца во флаконе прибавляют 1,0 мл воды Р и перемешивают до получения гомо-
генного раствора. Нагревают при температу-
ре 70°С в течение 45 мин.
Раствор сравнения. К 1,00 г испытуемого образца во флаконе прибавляют 50 мкл
этилен-оксида раствора Р4 и 950 мкл воды Р, плотно укупоривают. Нагревают при темпе-
ратуре 70°С в течение 45 мин. Условия хроматографирования:
–колонка кварцевая капиллярная длиной 30 м и диаметром 0,32 мм, покрытая слоем
поли(цианопропил)(7)(фенил)(7)(метил) (86)-силоксана Р (толщина слоя 1 мкм);
–газ-носитель: гелий для хроматографии Р;
–скорость подвижной фазы: 1,1 мл/мин.
–параметры парофазного пробоотбор-
ника:
–равновесная температура: 80°С,
– время |
достижения |
равновесия: |
45мин,
–температура передающей линии: 110°С,
–время приложения избыточного давления: 2 мин,
–время ввода пробы: 12 с;
– температура:
Трописетрона гидрохлорид |
593 |
|
Время (мин) |
Температура |
|
|
(°С) |
Колонка |
0—5 |
40 |
|
5—18 |
40 → 200 |
|
|
|
Блок ввода |
|
150 |
проб |
|
|
Детектор |
|
250 |
|
|
|
–детектор: пламенно-ионизационный;
–объем вводимой пробы: 1,0 мл.
Содержание этиленоксида Р в процентах
рассчитывают по формуле:
S m 10−4 |
, |
||
1 |
1 |
||
(S2 m2 ) − (S1 m3 ) |
|||
|
|||
где:
S1 — площадь пика этиленоксида на хро-
матограме испытуемого раствора;
S2 — площадь пика этиленоксида на хро-
матограме раствора сравнения;
m1 — масса навески этиленоксида, взято-
го для приготовления раствора сравнения, мкг; m2 — масса навески испытуемого образца, взятого для приготовления испытуемого
раствора, г;
m3 — масса навески испытуемого образца, взятого для приготовления раствора сравнения, г.
Тяжелые металлы (2.4.8, метод F). Не более 0,001 % (10 ppm). 1,0 г испытуемого об-
разца должен выдерживать испытание на тяжелые металлы. Эталон готовят с использо-
ванием 2 мл эталонного раствора свинца (10 ppm Pb) Р.
Потеря в массе при высушивании (2.2.32).
Не более 5,0%. 1,000 г испытуемого образца
сушат при температуре 105°С в течение 4 ч.
Сульфатная зола (2.4.14, метод А). Не более 0,4 %. Определение проводят из 1,0 г
испытуемого образца.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Троксерутин в условиях испытаний не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют
в 100,0 мл воды Р. 10,0 мл полученного раствора доводят водой Р до объема 100,0 мл. 10,0 мл полученного раствора доводят водой Р до объема 100,0 мл. Измеряют оптическую
плотность раствора (2.2.25) при 350 нм.
Содержание С33Н42О19 в процентах рас-
считывают с использованием удельного показателя поглощения, равного 250.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защищенном от света месте.
ТРОПИСЕТРОНА ГИДРОХЛОРИД
Tropisetroni hydrochloridum
TROPISETRON HYDROCHLORIDE
H |
|
|
O |
|
|
H |
|
HCl |
N |
CH3 |
|
O |
|
|
HN |
|
|
H |
|
|
С17Н20N2O2 · HCl |
М.м. 320,8 |
ОПРЕДЕЛЕНИЕ
Трописетрона гидрохлорид содержит не менее 99,0% и не более 101,0% (1R,3r,5S)-8-
метил-8-азабицикло[3.2.1]окт-3-ил]-1H-индол-
3-карбоксилата гидрохлорида в пересчете на
сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок. Легкорастворим или растворим в воде, уме-
ренно растворим в 96% спирте, очень мало растворим в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В, D. Вторая идентификация: А, С, D.
А. Абсорбционная спектрофотометрия в ультрафиолетовой и видимой областях (2.2.25).
Испытуемый раствор. 50 мг испытуемо-
го образца растворяют в метаноле Р и дово-
дят до объема 25,0 мл этим же растворителем. 1,0 мл полученного раствора доводят метанолом Р до 100,0 мл.
Диапазон длин волн: от 220 нм до 360 нм. Максимумы поглощения: при 228 нм и при
282нм.
Отношение оптических плотностей:
А228/А282 — от 1,3 до 1,4.
В. Абсорбционная спектрофотометрия в инфракрасной области (2.2.24).
Сравнение: ФСО трописетрона гидрохлорида # или спектр, представленный на рисунке 1.
C. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 5 мг испытуемого образца растворяют в смеси из равных объемов
метанола Р и метиленхлорида Р и доводят до объема 5 мл этой же смесью растворителей.
Раствор сравнения. 5 мг ФСО трописетрона гидрохлорида растворяют в смеси из равных объемов метанола Р и метиленхлорида Р и доводят до объема 5 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем силикагеля F254 Р.
594 |
Государственная фармакопея Республики Беларусь |
Пропускание
94
92
90
88
86
84
82
80
78
76
74
72
70
68
66
64
62
60
3000 |
2000 |
1500 |
1000 |
|
Волновое |
число (см-1) |
|
Рисунок 1. Инфракрасный спектр пропускания ФСО трописетрона гидрохлорида.
Подвижная фаза: кислота муравьиная безводная Р — вода Р — метанол Р — метиленхлорид Р (2:2:30:70, об/об/об/об).
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 2/3
высоты пластинки.
Высушивание: на холодном воздухе. Проявление А: пластинку просматрива-
ют в ультрафиолетовом свете при длине волны
254нм.
Результаты А: на хроматограмме испыту-
емого раствора обнаруживается пятно, соответствующее по расположению и размеру основному пятну на хроматограмме раствора сравнения.
Проявление В: пластинку опрыскивают раствором, приготовленным следующим образом: 0,85 г висмута нитрата основного Р растворяют в смеси из 10 мл кислоты уксусной Р и 40 мл воды Р; к 5 мл полученного раствора прибавляют 5 мл раствора 400 г/л калия йодида Р и доводят водой Р до объема 100 мл. Затем пластинку опрыскивают раствором пероксида водорода концентрированным Р.
Результаты В: на хроматограмме испытуемого раствора обнаруживается пятно, соответствующее по расположению и размеру основному пятну на хроматограмме раствора сравнения.
D. Испытуемый образец дает реакцию (а) на
хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор S. 1,00 г испытуемого образца растворяют в воде Р и доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор S должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора S должна быть не интенсивнее эталона В(К)7.
Примесь А. Не более 0,5%. Тонкослойная
хроматография (2.2.27).
Испытуемый раствор. 0,200 г испытуемого образца растворяют в смеси из равных объемов
метанола Р и метиленхлорида Р и доводят до
объема 5,0 мл этой же смесью растворителей.
Раствор сравнения (а). 5,0 мг ФСО тропина (примесь А) растворяют в смеси из равных
объемов метанола Р и метиленхлорида Р и доводят до объема 25,0 мл этой же смесью раст-
ворителей.
Раствор сравнения (b). 1,0 мл испытуемого раствора доводят смесью из равных объемов
метанола Р и метиленхлорида Р до объема 20,0 мл. К 0,1 мл полученного раствора прибавляют 1,0 мл раствора сравнения (а).
Пластинка: ТСХ пластинка со слоем сили-
кагеля F254 Р.
Подвижная фаза: раствор аммиака Р — метанол Р — метиленхлорид Р (5:40:60, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: не менее 2/3 высоты пластинки.
Высушивание: в потоке холодного воздуха.
Проявление: пластинку погружают в раствор калия йодовисмутата Р1.
Пригодность хроматографической системы: раствор сравнения (b):
– на хроматограмме обнаруживаются два полностью разделенных пятна.
Трописетрона гидрохлорид |
595 |
Предельное содержание примесей:
–примесь А: на хроматограмме испытуемо-
го раствора пятно, соответствующее примеси А,
должно быть не интенсивнее пятна на хроматограмме раствора сравнения (а).
Сопутствующие примеси. Жидкостная
хроматография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого образца растворяют в подвижной фазе А и
доводят до объема 20,0 мл этим же раствори-
телем.
Раствор сравнения (a). 1,0 мл испытуемого
раствора доводят подвижной фазой А до объема 100,0 мл. 1,0 мл полученного раствора доводят подвижной фазой А до объема 10,0 мл.
Раствор сравнения (b). 5,0 мг ФСО трописетрона примеси В и 5 мг ФСО этилиндол-3- карбоксилата растворяют в подвижной фазе А
идоводят до объема 20,0 мл этим же растворителем. 1,0 мл полученного раствора доводят подвижной фазой А до объема 50,0 мл.
Условия хроматографирования:
–колонка длиной 0,25 м и внутренним диаметром 4,6 мм, заполненная силикагелем октилсилильным для хроматографии Р с размером частиц 5 мкм;
–подвижная фаза:
–подвижная фаза А: триэтиламин Р — ацетонитрил Р — вода Р — метанол Р
(0,3:35:400:565, об/об/об/об);
–подвижная фаза В: триэтиламин Р — ацетонитрил Р — вода Р — метанол Р
(0,3:100:100:800, об/об/об/об);
Время (мин) |
Подвижная |
Подвижная |
фаза А |
фаза В |
|
|
(%, об/об) |
(%, об/об) |
0—14 |
100 |
0 |
14—32 |
100 → 0 |
0 → 100 |
32—36 |
0 |
100 |
36—37 |
0 → 100 |
100 → 0 |
37—52 |
100 |
0 |
–скорость подвижной фазы: 2 мл/мин;
–спектрофотометрический детектор,
длина волны 280 нм;
–объем вводимой пробы: 20 мкл.
Относительное удерживание (по отношению
ктрописетрону; время удерживания — около
22 мин): примесь В — около 0,05; этилиндол-3-
карбоксилат — около 0,2.
Пригодность хроматографической системы: раствор сравнения (b):
–разрешение: не менее 4 между пиком при-
меси В и пиком этилиндол-3-карбоксилата.
Предельное содержание примесей:
–примесь В (не более 0,1%): на хроматограмме испытуемого раствора площадь пика, соответствующего примеси В, не должна превышать площадь соответствующего пика на хрома-
тограмме раствора сравнения (b);
–неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раст-
вора площадь пика, соответствующего любой
неспецифицированной примеси, не должна пре-
вышать площадь основного пика на хроматограмме раствора сравнения (а);
–сумма примесей (не более 0,3%): на хро-
матограмме испытуемого раствора сумма площадей пиков, соответствующих всем примесям, не должна превышать 3-кратную площадь основного пика на хроматограмме раствора сравнения (а).
На хроматограмме испытуемого раствора
не учитывают пики с площадью менее 0,5 площади основного пика на хроматограмме раст-
вора сравнения (а) (0,05%).
N,N-Диметиланилин. Не более 0,002% (20 ppm). Жидкостная хроматография (2.2.29).
Растворы готовят непосредственно перед использованием.
Испытуемый раствор. 250 мг испытуемо-
го образца растворяют в подвижной фазе А и доводят до объема 5,0 мл этим же раствори-
телем.
Раствор сравнения. 10,0 мг N,N-диметил- анилина Р растворяют в подвижной фазой А и
доводят до объема 100,0 мл этим же растворителем. 1,0 мл полученного раствора доводят подвижной фазой А до объема 100,0 мл.
Условия хроматографирования:
–колонка длиной 0,25 м и внутренним диаметром 4,6 мм, заполненная силикагелем октилсилильным для хроматографии Р с размером частиц 5 мкм;
–подвижная фаза:
–подвижная фаза А: триэтиламин Р — ацетонитрил Р — вода Р — метанол Р
(0,3:35:400:565, об/об/об/об);
–подвижная фаза В: триэтиламин Р — ацетонитрил Р — вода Р — метанол Р
(0,3:100:100:800, об/об/об/об/об);
Время (мин) |
Подвижная |
Подвижная |
фаза А |
фаза В |
|
|
(%, об/об) |
(%, об/об) |
0—10 |
100 |
0 |
10—11 |
100 → 0 |
0 → 100 |
11—30 |
0 |
100 |
30—31 |
0 → 100 |
100 → 0 |
31—50 |
100 |
0 |
–скорость подвижной фазы: 1 мл/мин;
–спектрофотометрический детектор, длина волны 248 нм;
–объем вводимой пробы: 20 мкл. Предельное содержание примесей:
–N,N-диметиланилин: на хроматограмме испытуемого раствора площадь пика N,N- диметиланилина не должна превышать площадь основного пика на хроматограмме раствора сравнения.
596 |
Государственная фармакопея Республики Беларусь |
Потеря в массе при высушивании (2.2.32).
Не более 0,3%. 1,000 г испытуемого образца
сушат при температуре 105°С.
Сульфатная зола (2.4.14, метод А). Не более 0,1%. Определение проводят из 1,0 г ис-
пытуемого образца.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец
должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Трописетрона гидрохлорид в усло-
виях испытания обладает антимикробным дей-
ствием. Посев на питательные среды № 1, № 8,
№11 проводят из разведения 1:100, на питательную среду № 2 — из разведения 1:10.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,250 г испытуемого образца растворяют в
10 мл кислоты уксусной безводной Р, прибавляют 70 мл уксусного ангидрида Р и титруют 0,1 М раствором кислоты хлорной потенциометриче-
ски (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соот-
ветствует 32,08 мг С17Н20N2O2·HCl.
ПРИМЕСИ
Специфицированные примеси: A, B.
H
H |
|
N |
CH3 |
HO |
|
H |
|
А. (1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан- 3-ол (тропин).
CO2H
HN
В. 1H-Индол-3-карбоновая кислота.
ФАМОТИДИН
Famotidinum
FAMOTIDINE
|
|
NH2 |
NH2 |
|
||||
|
|
|
|
|||||
H2N |
|
|
|
N |
|
|
||
|
|
|
|
|
||||
|
|
|
|
|
|
S |
N |
NH2 |
|
|
|
|
|
|
|||
|
|
N |
|
|
|
|
|
S |
|
|
|
|
|
||||
|
|
|
|
S |
O |
O |
||
C8H15N7O2S3 |
М.м. 337,5 |
|||||||
|
ОПРЕДЕЛЕНИЕ |
|
|
|||||
|
Фамотидин содержит |
не менее |
98,5% |
|||||
и не более 101,5% 3-[[[2-[(диаминомети- лен)амино]тиазол-4-ил]метил]сульфанил]-N’- сульфамоилпропанимидамида в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый кристаллический порошок либо кристаллы.
Очень мало растворим в воде, легкорас-
творим в кислоте уксусной ледяной, очень мало
растворим в этаноле, практически нерастворим
в этилацетате. Растворяется в разведенных минеральных кислотах.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в ин-
фракрасной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО фамотидина # или спектр,
представленный на рисунке 1.
Если полученные спектры отличаются, то 0,10 г испытуемого образца и 0,10 г ФСО фамотидина суспендируют по отдельности в 5 мл
воды Р, нагревают до кипения и охлаждают, по-
тирая стенки пробирки стеклянной палочкой для инициации кристаллизации. Фильтруют, промы-
вают кристаллы 2 мл ледяной воды Р и сушат при температуре 80°С и давлении, не превыша-
ющем 670 Па, в течение 1 ч. Сухие остатки ис-
пользуют для получения новых спектров.
ИСПЫТАНИЯ
Раствор S. 0,20 г испытуемого образца
растворяют в растворе 50 г/л кислоты хлористоводородной Р, нагревая при необходимости до 40°С, и доводят до объема 20 мл этим же растворителем.
Прозрачность (2.2.1). Раствор S должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора S должна быть не интенсивнее эталона BY(КЖ)7.
Сопутствующие примеси. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 12,5 мг испытуемого
образца растворяют в подвижной фазе А и дово-
дят до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раствора доводят подвижной фазой А до объема 10,0 мл. 1,0 мл полученного раствора доводят
подвижной фазой А до объема 100,0 мл.
Раствор сравнения (b). 2,5 мг ФСО фамотидина примеси D растворяют в метаноле Р и
доводят до объема 10,0 мл этим же растворите-
лем. К 1,0 мл полученного раствора прибавляют 0,50 мл испытуемого раствора и доводят подвижной фазой А до объема 100,0 мл.
Раствор сравнения (с). 5,0 мг ФСО фамотидина для проверки пригодности хроматографической системы (содержит примеси A,
B, C, D, E, F, G) растворяют в подвижной фазе А и доводят до объема 10,0 мл этим же растворителем.
Условия хроматографирования:
– колонка длиной 0,25 м и внутренним диаметром 4,6 мм, заполненная силикагелем окта-
Фамотидин |
597 |
Пропускание
95
90
85
80
75
70
65
60
55
50
45
40
35
30
25
20
15
10
3000 |
2000 |
1500 |
1000 |
500 |
Волновое число (см-1)
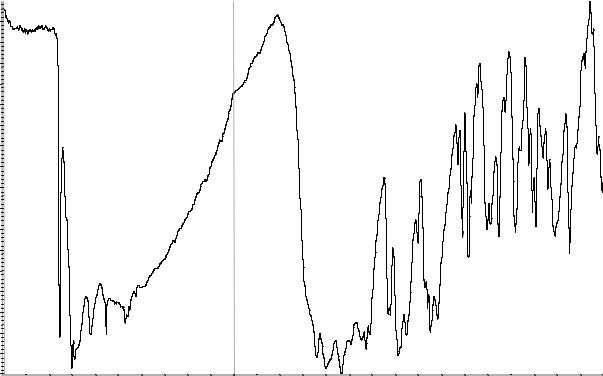
Рисунок 1. Инфракрасный спектр пропускания ФСО фамотидина в дисках с калия бромидом Р.
децилсилильным эндкепированным для хроматографии Р с размером частиц 5 мкм;
–температура: 50°С;
–подвижная фаза:
–подвижная фаза А: смесь из 6 частей метанола Р, 94 частей ацетонитрила Р и 900 объемов раствора 1,882 г/л натрия гексансульфоната Р, доведенного кислотой уксусной Р до рН 3,5;
–подвижная фаза В: ацетонитрил Р;
Время |
Подвиж- |
Подвиж- |
Скорость |
ная фаза |
ная фаза |
подвижной |
|
(мин) |
А |
В |
фазы |
|
(%, об/об) |
(%, об/об) |
(мл/мин) |
0—23 |
100 → 96 |
0 → 4 |
1 |
23—27 |
96 |
4 |
1 → 2 |
27—47 |
96 → 78 |
4 → 22 |
2 |
47—48 |
78 → 100 |
22 → 0 |
2 |
48—54 |
100 |
0 |
2 → 1 |
|
|
|
|
–спектрофотометрический детектор, длина волны 265 нм;
–объем вводимой пробы: 20 мкл. Относительные времена удерживания (по
отношению к фамотидину; время удержива-
ния — около 21 мин): примесь D — около 1,1; примесь С — около 1,2; примесь G — около 1,4; примесь F — около 1,5; примесь А — около 1,6; примесь В — около 2,0; примесь Е — около 2,1.
Пригодность хроматографической системы:
–хроматограмма раствора сравнения (с)
должна соответствовать хроматограмме ФСО фамотидина для проверки пригодности хроматографической системы;
–времена удерживания: фамотидин — 19—23 мин на всех хроматограммах; примесь
Е— не более 48 мин на хроматограмме раствора сравнения (с);
–разрешение: не менее 3,5 между пиками фамотидина и примеси D на хроматограмме раствора сравнения (b).
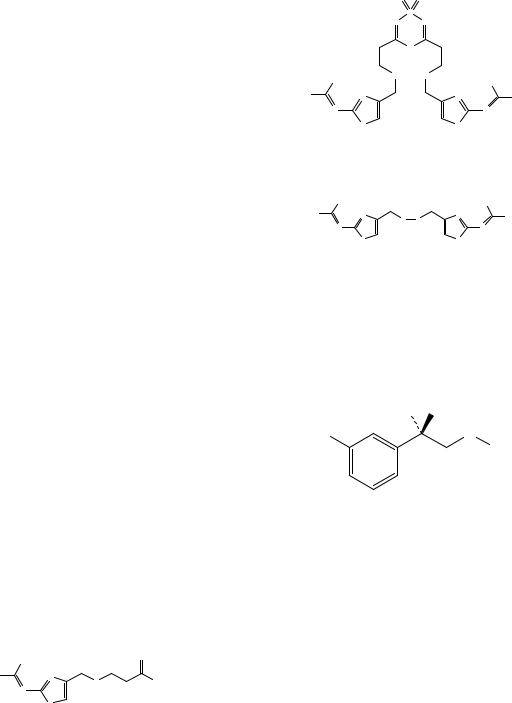
Предельное содержание примесей (для расчета содержания примесей умножают площади пиков на соответствующие поправочные коэффициенты: для примеси А — 1,9; для примеси В — 2,5; для примеси С — 1,9; для примеси F — 1,7; для примеси G — 1,4):
–примеси А, G (не более 0,2%): на хроматограмме испытуемого раствора площадь пика, соответствующего примеси, не должна превы-
шать 2-кратную площадь основного пика на хро-
матограмме раствора сравнения (а);
–примеси B, C, D, E (не более 0,3 %): на хроматограмме испытуемого раствора площадь
каждого пика, соответствующего примеси, не
должна превышать 3-кратную площадь основ-
ного пика на хроматограмме раствора сравнения (а), и не более трех из них могут иметь
площадь, превышающую площадь основного пика на хроматограмме раствора сравнения
(а) (0,1 %);
–примесь F (не более 0,1%): на хроматограмме испытуемого раствора площадь пика,
соответствующего примеси F, не должна превы-
598 |
Государственная фармакопея Республики Беларусь |
шать площадь основного пика на хроматограм-
ме раствора сравнения (а);
–любая другая примесь (не более 0,1% и
не более 0,05%): на хроматограмме испытуемого раствора площадь любого пика, кроме пика
фамотидина и пиков примесей А, В, С, D, Е, F, G,
не должна превышать площадь основного пика на хроматограмме раствора сравнения (а) для
пиков со временем выхода менее 25 мин (0,1%)
ине должна превышать 0,5 площади основно-
го пика на хроматограмме раствора сравнения
(а) для пиков со временем выхода более 25 мин (0,05%);
–сумма примесей (не более 1,0%): на хроматограмме испытуемого раствора сумма пло-
щадей всех пиков, кроме основного, не должна превышать 10-кратной площади основного пика
на хроматограмме раствора сравнения (а).
На хроматограмме испытуемого раствора
не учитывают пики с площадью менее 0,2 пло-
щади основного пика на хроматограмме раст-
вора сравнения (а) (0,02%).
Тяжелые металлы (2.4.8, метод D). Не
более 0,001% (10 ppm). 2,0 г испытуемого об-
разца должны выдерживать испытание на тя-
желые металлы. Эталон готовят с использованием 2 мл эталонного раствора свинца (10 ppm Pb) P.
Потеря в массе при высушивании
(2.2.32). Не более 0,5%. 1,000 г испытуемого об-
разца сушат при температуре 80°С и давлении, не превышающем 670 Па, в течение 5 ч.
Сульфатная зола (2.4.14, метод А). Не более 0,1%. Определение проводят из 1,0 г ис-
пытуемого образца.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Фамотидин в условиях испытания
не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,120 г испытуемого образца растворяют в
60 мл кислоты уксусной безводной Р и титруют
0,1 М раствором хлорной кислоты потенциометрически (2.2.20).
1 мл 0,1 М раствора хлорной кислоты соот-
ветствует 16,87 мг С8Н15N7O2S3.
ХРАНЕНИЕ
В защищенном от света месте.
ПРИМЕСИ
|
NH2 |
X |
|
|
|
H2N |
N |
R |
|
S |
|
|
N |
|
|
S |
|
А. R = NН2, Х = NH: 3-[[[2-[(Диаминометилен)- амино]тиазол-4-ил]метил]cуль-фанил]пропани- мидамид.
C.R = NH-SO2-NH2, X = O: 3-[[[2-[(Диамино-
метилен)амино]тиазол-4-ил]метил]сульфанил]-
N-сульфамоилпропанамид.
D.R = NН2, Х = О: 3-[[[2-[(Диаминометилен)-
амино]тиазол-4-ил]метил]суль-фанил]пропана-
мид.
F.R = ОН, Х = О: 3-[[[2-[(Диаминометилен)-
амино]тиазол-4-ил]метил]суль-фанил]пропионо- вая кислота.
G.R = NH-CN, X = NH: N-Циано-3-[[[2-
[(диаминометилен)амино]тиазол-4-ил]метил]-
сульфанил]пропанимидамид.
OO
S
NN
N
H
|
S |
S |
H2N |
|
NH2 |
|
|
H2N |
N |
N |
NH2 |
|
N |
|
N |
SS
В.3,5-бис[2-[[[2-[(Диаминометилен)амино]-
тиазол-4-ил]метил]сульфанил]этил]-4Н-1,2,4,6- тиатриазина 1,1-диоксид.
|
NH2 |
|
H2N |
H2N |
N |
N |
NH2 |
|
S |
S |
|
|
N |
|
N |
SS
Е.2,2’-[Дисульфандиилбис(метилентиазол- 4,2-диил)]дигуанидин.
ФЕНИЛЭФРИН
Phenylephrinum
PHENYLEPHRINE
HOH
HO |
H |
N |
|
|
CH3 |
C9H13NO2 |
М.м. 167,2 |
ОПРЕДЕЛЕНИЕ
Фенилэфрин содержит не менее 99,0 % и не более 100,5 % (1R)-1-(3-гидроксифенил)-2- (метиламино)этанола в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Малорастворим в воде, умеренно растворим в метаноле, малорастворим в 96 % спирте. Растворяется в разведенных минеральных кислотах и разведенных растворах гидроксидов щелочных металлов.
Температура плавления: около 174°С.
Фенилэфрин |
599 |
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В. Вторая идентификация: A, С, D.
А. Испытуемый образец выдерживает испытание «Удельное оптическое вращение» как указано в разделе «Испытания».
В. Абсорбционная спектрофотометрия в ин-
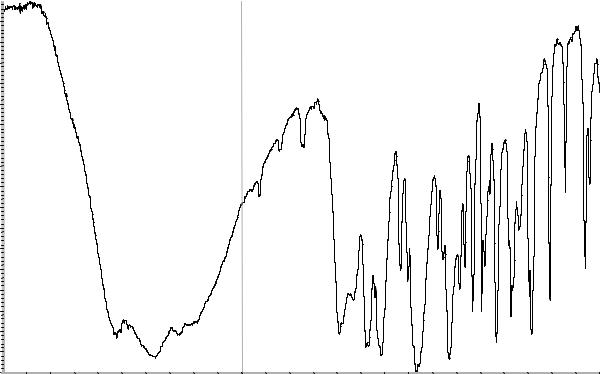
фракрасной области (2.2.24).
Сравнение: ФСО фенилэфрина # или спектр, представленный на рисунке 1.
С. Тонкослойная хроматография (2.2.27).
Смесь растворителей. Смесь из равных
объемов метиленхлорида Р и метанольного раствора кислоты хлористоводородной (1 объем
кислоты хлористоводородной Р доводят метанолом Р до 10 объемов).
Испытуемый раствор. 0,1 г испытуемого образца растворяют в смеси растворителей и доводят до объема 5 мл этим же растворителем.
Раствор сравнения. 20 мг ФСО фенилэфрина растворяют в смеси растворителей и дово-
дят до объема 1 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем сили-
кагеля F254 Р.
Подвижная фаза: раствор аммиака концентрированный Р — метанол Р — метиленхлорид Р (0,5:25:70, об/об/об).
Наносимый объем пробы: 10 мкл.
Фронт подвижной фазы: на менее 15 см от линии старта.
Высушивание: в потоке холодного воздуха. Проявление: пластинку просматривают в уль-
трафиолетовом свете при длине волны 254 нм; пластинку опрыскивают раствором 1 г/л соли проч-
ного синего В Р в растворе 50 г/л натрия карбоната Р и просматривают при дневном свете.
Результаты: на хроматограмме испытуемого раствора обнаруживается основное пятно,
соответствующее по расположению, цвету и раз-
меру основному пятну на хроматограмме раст-
вора сравнения.
D.10 мг испытуемого образца растворяют
в1 мл 1 М растворе кислоты хлористоводородной, прибавляют 0,05 мл раствора меди сульфата Р и 1 мл раствора 200 г/л натрия гидроксида Р. Появляется фиолетовое окрашивание. Прибавляют 1 мл эфира Р и встря-
хивают. Верхний слой остается бесцветным.
ИСПЫТАНИЯ
Раствор S. 1 г испытуемого образца раст-
воряют в 1 М растворе кислоты хлористоводородной и доводят до объема 10 мл этим же растворителем.
Прозрачность (2.2.1). Раствор S должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска
раствора S должна быть не интенсивнее эталона Y(Ж)7.
Удельное оптическое вращение (2.2.7).
От -53 до -57 в пересчете на сухое вещество.
1,250 г испытуемого образца растворяют в
1 М растворе кислоты хлористоводородной
и доводят до объема 25,0 мл этим же раство-
рителем.
Сопутствующие примеси. Жидкостная хроматография (2.2.29).
Смесь растворителей. Раствор кислоты хлористоводородной разведенной Р —
Пропускание
95
90
85
80
75
70
65
60
55
50
45
40
35
30
25
20
15
10
3000 |
2000 |
1500 |
1000 |
|
Волновое |
число (см-1) |
|
Рисунок 1. Инфракрасный спектр пропускания ФСО фенилэфрина в дисках с калия бромидом Р.
600 |
Государственная фармакопея Республики Беларусь |
подвижная фаза В — подвижная фаза А
(5:200:800, об/об/об).
Буферный раствор рН 2,8. 3,25 г натрия октансульфоната Р растворяют в 1000 мл воды Р при перемешивании в течение 30 мин и доводят кислотой фосфорной разведенной Р
до рН 2,8.
Испытуемый раствор. 41,0 мг испытуемого образца растворяют в смеси растворителей и до-
водят до объема 50,0 мл этим же растворителем.
Раствор сравнения (а). 5,0 мл испытуе-
мого раствора доводят смесью растворителей
до объема 100,0 мл. 2,0 мл полученного раствора доводят смесью растворителей до объема 100,0 мл.
Раствор сравнения (b). Содержимое контейнера с ФСО фенилэфрина гидрохлорида для идентификации пиков (содержит примеси С и Е) растворяют в 2 мл смеси растворителей.
Условия хроматографирования:
–колонка длиной 0,055 м и внутренним диа-
метром 4,0 мм, заполненная силикагелем октадецилсилильным эндкепированным для хроматографии Р с размером частиц 3 мкм;
–температура: 45°С;
–спектрофотометрический детектор,
длина волны 215 нм;
–подвижная фаза:
–подвижная фаза А: ацетонитрил Р1 — буферный раствор рН 2,8 (10:90, об/об);
–подвижная фаза В: буферный раствор
рН 2,8 — ацетонитрил Р1 (10:90, об/об);
Время (мин) |
Подвижная |
Подвижная |
фаза А |
фаза В |
|
|
(%, об/об) |
(%, об/об) |
0—3 |
93 |
7 |
3—13 |
93 → 70 |
7 → 30 |
13—14 |
70 → 93 |
30 → 7 |
|
|
|
–скорость подвижной фазы: 1,5 мл/мин;
–объем вводимой пробы: 10 мкл. Относительное удерживание (по отно-
шению к фенилэфрину; время удерживания —
около 2,8 мин): примесь С — около 1,3; примесь
Е — около 3,6.
Пригодность хроматографической системы:
–фактор асимметрии: не более 1,9 для
основного пика на хроматограмме испытуемого
раствора;
–коэффициент разделения пиков: не
менее 5 (Hp — высота пика примеси С относительно базовой линии; Hv — расстояние между базовой линией и нижней точкой кривой, разде-
ляющей пики примеси С и фенилэфрина).
Предельное содержание примесей (для расчета содержания примесей умножают площади пиков на соответствующие поправочные коэффициенты: для примеси С — 0,5, для примеси Е — 0,5):
–примеси С, Е (не более 0,1%): на хрома-
тограмме испытуемого раствора площади пиков, соответствующих примесям С и Е, не должны
превышать площадь основного пика на хромато-
грамме раствора сравнения (а);
–неспецифицированные примеси (не более
0,10%): на хроматограмме испытуемого раст-
вора площадь пика, соответствующего любой неспецифицированной примеси, не должна пре-
вышать площадь основного пика на хромато-
грамме раствора сравнения (а);
–сумма примесей (не более 0,2%): на хро-
матограмме испытуемого раствора сумма пло-
щадей пиков, соответствующих всем примесям, не должна превышать 2-кратную площадь
основного пика на хроматограмме раствора
сравнения (а).
На хроматограмме испытуемого раствора не учитывают пики с площадью менее 0,5 площади основного пика на хроматограмме раствора сравнения (а) (0,05%).
Потеря в массе при высушивании (2.2.32).
Не более 0,5%. 1,000 г испытуемого образца
сушат при температуре 105°С.
Сульфатная зола (2.4.14, метод А). Не более 0,1%. Определение проводят из 1,0 г ис-
пытуемого образца.
# Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
# Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Фенилэфрин в условиях испытания не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в
60 мл уксусной кислоты безводной Р и титруют
0,1 М раствором кислоты хлорной потенциоме-
трически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соответствует 16,72 мг C9H13NO2.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: С, Е. Другие обнаруживаемые примеси (сле-
дующие вещества, если они присутствуют в значительных количествах, следует определять тем или иным испытанием, описанным в частной статье. Их содержание лимитируется общим критерием приемлемости для других/ неспецифицированных примесей и/или общей статьей Субстанции для фармацевтического использования. Вследствие этого нет необходимости идентифицировать эти примеси для доказательства соответствия требованиям. См. также статью 5.10 Контроль примесей в субстанциях для фармацевтического использования): А, D.