

PRKT
.pdfгде Е20 – ЭДС нормального элемента при 20 °С, приводимая в паспорте данного экземпляра нормального элемента. Полученное значение установить с помощью регуляторов 5 потенциометра. При установленном в положение «1» переключателе 3 переключатель рода работ 2 установить в положение «IA», а кнопку 1 «Измерение» нажать и повернуть. При этом она должна зафиксироваться в нижнем положении. Поворачивая переключатель 3 по часовой стрелке на одно деление (увеличивая чувствительность прибора), следить за зайчиком гальванометра. При некотором положении переключателя 3 штрих зайчика отклонится от нуля. Вращая маленькие ручки «Тонко» ряда «IA», установить штрих на нуль1 и снова увеличить чувствительность. Если при этом зайчик опять отклонится, вернуть его на нуль, вращая те же ручки «Тонко». Операция повторяется до тех пор, пока при положении переключателя 3 на цифре «6» штрих гальванометра не установится на нулевое деление.
Снова установить минимальную чувствительность прибора (на цифре «1») и приступить регулировке рабочего тока IB, для чего переключатель 2 перевести в положение «IB» и выполнить те же операции, что и при установке тока IA, используя ряд ручек «IB». Аналогично, если это необходимо, установить рабочий ток IС, пользуясь для регулировки отвёрткой. Переключатель 3 перевести в положение «1». Прибор готов к работе.
Переключатель 2 установить в положение «Х1» или «Х2», в зависимости от клемм, к которым подключена исследуемая электрохимическая цепь. Зайчик гальванометра отклонится от нулевого положения. Компенсацию ЭДС производить переключателями декад «U1» или «U2» (индекс совпадает с индексом клемм, к которым подключена цепь). Вращением первого переключателя слева подвести штрих гальванометра к нулевой отметке, увеличить чувствительность на одно деление (в положение «2») и снова тем же переключателем декады подвести штрих к нулю. Увеличить чувствительность и переключателем второй декады опять установить штрих на нуль. Увеличивая чувствительность и вращая переключатели декад, перемещаясь в их ряду слева направо, добиться такого положения, чтобы при шестом положении переключателя 3 штрих гальванометра находился на нулевой отметке. В окошках под переключателями отсчитать величину ЭДС.
Если в течение полутора – двух минут колебание показания потенциометра не будет выходить из диапазона, не превышающего ±0,01 мВ, можно перейти к измерению ЭДС при другой температуре. В противном случае необходимо дождаться установление теплового равновесия в системе термостат – электрохимическая цепь и постоянства ЭДС.
Переключатель 3 перевести в положение «1». Вращая головку контактного термометра, установить указатель на его верхней шкале на сле-
1 Если маленькими ручками это сделать не удаётся, произвести установку большими ручками “Грубо” этого же ряда.
31

дующее, более высокое, заданное значение температуры. Через 15 – 20 минут измерить ЭДС, как описано выше.
После завершения всех измерений освободить кнопку 1 «Измерение», повернув её. Остальные органы управления перевести в исходные положения. Выключить все приборы.
Построить график зависимости Е от Т, выбрав такими его размеры и масштаб, чтобы при построении зависимости не округлять измеренные значения ЭДС и температуры. В узком диапазоне температур графическая зависимость Е от Т практически линейная1, поэтому dE/dT – тангенс угла наклона полученной прямой к оси температур. Результаты измерений и вычислений свести в таблицу.
Т |
Е |
dE/dT |
∆G |
∆H |
∆S |
W |
|
|
|
|
|
|
|
Вопросы для самопроверки
1.Почему ЭДС, являясь мерой работы по обратимому переносу заряда по внешней цепи, выражает величину ∆G реакции, идущей в гальваническом элементе?
2.Почему величина ЭДС зависит от температуры?
3.В каком случае ЭДС увеличивается с ростом температуры?
4.Выведите уравнения (1.9) – (1.13).
5.Почему термодинамические параметры реакции, проводимой по электрохимическому механизму, определённые методом ЭДС, применимы
креакции в любых условиях?
Задачи
1.Температурный коэффициент ЭДС элемента, в котором идёт двухэлектронная реакция, равен –3,7 10–4 В К–1. Вычислить теплоту при обратимой работе элемента.
2.Элемент, образованный иодсеребряным и иодсвинцовым электродами, при 298 К имеет ЭДС, равную 0,2169 В. Температурный коэффициент
ЭДС равен 1,38 10–4 В К–1. Изобразить схему элемента, записать уравнение реакции в элементе и рассчитать Е, ∆G, ∆Н, ∆S и W при 310 К. Зависит ли величина Е от концентрации иодида в элементе? Почему?
3. Стандартная ЭДС элемента
Pt, H2 | HCl(p-p) | Hg2Cl2(тв) | Hg
при 20 °С равна 0,2692 В, и 0,2660 В при 30 °С. Запишите уравнение реакции в элементе, вычислите для неё ∆G°, ∆Н°, ∆S° , а также W° при обратимой работе элемента.
1 Если график строится с помощью компьютера, следует зависимость аппроксимировать уравнением прямой E = a +bT, в котором константа b равна температурному коэффициенту ЭДС.
32
2. ХИМИЧЕСКАЯ КИНЕТИКА
ФОРМАЛЬНАЯ КИНЕТИКА
Краткое теоретическое введение
Если реагенты (или реагенты и катализатор) находятся в различных фазах, то реакция называется гетерогенной; Если же реакция протекает в одной фазе, то она называется гомогенной. Гомогенные реакции идут в объёме фазы, гетерогенные – на поверхности раздела фаз.
Скорость гомогенных реакций определяется изменением молярной концентрации с в единицу времени t и выражается как производная концентрации по времени для любого вещества, участвующего в реакции, как
вкачестве исходного компонента, так и в качестве продукта.
Вобщем случае скорость реакции, протекающей по уравнению
′ ′ |
(2.А) |
ν1 A +ν2B ↔ν1C +ν2D , |
где ν1, ν2, ν1′, ν2′ – стехиометрические коэффициенты, может быть представленанесколькимивыражениями:
w=− |
1 dcA |
=− |
1 dcB |
= |
|
1 dcC |
= |
|
1 dcD |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
. |
(2.1) |
||||
ν dt |
ν dt |
ν |
' |
|
dt |
ν |
' |
dt |
|||||||||||||
|
1 |
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
1 |
|
|
|
2 |
|
|
|
|
||||
Исходные вещества расходуются, а продукты реакции образуются в эквивалентных количествах (соответственно стехиометрическим коэффициентам), поэтому при определении скорости реакции нет необходимости следить за изменением концентрации всех взаимодействующих веществ.
Скорость реакции можно определить и как производную степени полноты реакции (глубина ее протекания) по времени. Скорость реакции зависит от природы реагирующих веществ, их концентрации, присутствия посторонних веществ (например, катализаторов) и их концентрации. На скорость реакции влияют также среда, в которой протекает реакция, и условия протекания реакции: температура, давление (особенно для реакций с участием газов), облучение (фотохимические реакции) и т. п.
Основным законом химической кинетики является постулат, выражающий зависимость скорости реакции от концентрации реагирующих веществ. В соответствии с ним скорость реакции в каждый момент времени пропорциональна произведению возведенных в некоторую степень концентраций реагирующих веществ (закон действия масс). Так, для реакции (2.А) скорость, равная разности скоростей прямой и обратной реакций, может быть выражена кинетическим уравнением
w = k1[A]n1 [B]n2 − k2[C]n3 [D]n4 |
(2.2) |
Коэффициенты пропорциональности k1 и k2 в уравнении (2.2) называются константой скорости реакции (прямой и обратной).
33

Когда k1 >> k2 , т.е. скорость обратной реакции ничтожно мала по сравнению со скоростью прямой, равновесие оказывается полностью сдвинутым вправо и уравнение (2.2) переходит в уравнение
− |
dc |
= k[A]n1 [B]n2 |
(2.3) |
|
dt |
||||
|
|
|
Из уравнений (2.2) и (2.3) видно, что скорость необратимой1 (односторонней) реакции зависит только от концентрации исходных веществ, в случае обратимой реакции (2.А) – от концентраций как исходных веществ. так и продуктов реакции.
Константа скорости k в уравнении (2.3) равняется скорости реакции при условии, что концентрации каждого из реагирующих веществ равны единице, поэтому её называют также удельной скоростью реакции. Такой физический смысл константы скорости указывает на то, что величина ее должна зависеть от всех факторов, которые влияют на скорость реакции, за исключением концентрации реагирующих веществ. Численное значение константы скорости зависит также от выбора единиц времени и концентрации. Размерность константы определяется тем кинетическим уравнением, по которому производится её расчет, т. е. зависит от порядка реакции.
Каждая реакция характеризуется порядком, а также её молекулярностью. Молекулярность реакции определяется числом частиц, участвующих в одном элементарном акте химического превращения. При этом число молекул продуктов процесса не имеет значения. В зависимости от этого различаютреакции: 1) мономолекулярные, 2) бимолекулярные, 3) тримолекулярные.
Порядок реакции определяется показателем степени при концентрации в кинетическом уравнении реакции. Если порядок равен единице, то реакцию называют реакцией первого порядка, если двум – второго порядка, если трём – третьего порядка. В зависимости от порядка реакции кинетические уравнения для расчета константы скорости реакции различны.
Порядок реакции является чисто эмпирической величиной. Только для элементарной реакции, протекающей в один этап, он равен её молекулярности, так как стехиометрическое уравнение правильно отражает истинный механизм лишь такой реакции. Различают полный и частный порядок реакции. Каждый из показателей степени при концентрациях в дифференциальном уравнении скорости выражает частный порядок по данному веществу. Сумма показателей степени при концентрациях определяет полный (суммарный) порядок реакции.
Протекание реакции сложным путем, в несколько стадий, является одной из причин расхождения между молекулярностью и порядком реакции. Другой причиной расхождения может быть значительный избыток одного из реагентов в реакционной смеси. Тогда концентрация этого реагента остаётся практически постоянной в ходе реакции, а порядок реакции будет меньше,
1 Не путать с термодинамической необратимостью!
34
чем определяемый по стехиометрическому уравнению. Например, такие бимолекулярные реакции, как инверсия тростникового сахара или гидролиз уксусного ангидрида оказываются кинетически реакциями первого порядка, так как концентрацию воды здесь можно считать неизменной. Подобного рода реакции иногда называют псевдомономолекулярными. Порядок реакции зависит от условий её протекания. Его можно изменить, например, варьированиемконцентрации или давления.
Кинетическая классификация реакций. В химической кинетике реакции разделяют по следующим признакам:
1)По числу частиц, участвующих в реакции (молекулярность и порядок реакции);
2)По природе частиц, участвующих в элементарном акте реакции. Реакции, в которых участвуют молекулы, относятся к группе молекулярных реакций; реакции с участием атомов или свободных радикалов – к группе цепных реакций; реакции с участием ионов – кгруппеионныхреакций;
3)По числу фаз, участвующих в реакции. Реакции, протекающие в одной фазе, называют гомогенными. Реакции, протекающие на поверхности или
уповерхностиразделафаз, называютгетерогенными;
4)По использованию катализаторов: каталитические, автокаталитическиеинекаталитические;
5)По степени сложности (по механизму протекания): a) обратимые и необратимые; б) изолированные и параллельные; в) последовательные; г) сопряжённые.
Большинство реакций моно- и бимолекулярные. Вероятность одновременного столкновения большего числа молекул определённого вида ничтожно мала; поэтому даже тримолекулярные реакции немногочисленны. По числу стадий реакции делятся на простые и сложные. Простая (элементарная) реакция состоит из однотипных элементарных актов. В простой химической реакции реагенты непосредственно превращаются в конечные продукты без образования промежуточных веществ. Простая реакция протекает без участия катализатора и не тормозится ингибиторами. Для простой реакции, как правило, существует лишь одно переходное состояние.
Сложная (многостадийная) реакция состоит из нескольких (иногда большого числа) простых реакций – элементарных стадий, связанных друг с другом через реагенты или продукты. Часто продукт одной стадии служит реагентом для других стадий, являясь промежуточным продуктом. Если промежуточный продукт очень быстро подвергается последующим превращениям, то он является лабильным продуктом и обычно присутствует в системах в весьма низкой квазистационарной концентрации.
Следовательно, если стехиометрические коэффициенты в уравнении химической реакции велики, то реакция протекает в несколько стадий и общая скорость реакции определяется скоростью самой медленной из них.
35
В зависимости от уравнения, связывающего скорость реакция с концентрацией реагирующих веществ, различают реакции первого порядка (сумма показателей степени в уравнении (2.3) равна 1), второго порядка и т.д. Порядок реакции может быть отрицательным, нулевым, дробным. Если порядок реакции не целочисленный и не равен молекулярности реакции, то изучаемая реакция относится к сложным.
Реакция первого порядка схематически может быть представлена уравнением
A → B (возможно образование нескольких продуктов). Её скорость определяется дифференциальным уравнением
− dc |
= kcA = k(cA0 |
−cx ) , |
(2.4) |
dt |
|
|
|
которое после интегрирования даёт
k = |
1 |
ln |
c0 |
= |
1 |
ln |
|
c0 |
= − |
1 |
ln(1 |
−α) , |
|
||||||
|
|
|
A |
|
|
|
|
A |
|
|
|
(2.5) |
|||||||
|
t |
cA |
|
t |
0 |
|
|
t |
|||||||||||
|
|
|
|
|
|
cA −cx |
|
|
|
|
|
||||||||
а период полураспада определяется как τ1/ 2 = ln 2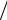 k = 0.69315k −1 , где cA0 – начальная концентрация вещества А, cx – концентрация прореагировавшего вещества А за истекший промежуток времени; cA = cA0 − cx – концен-
k = 0.69315k −1 , где cA0 – начальная концентрация вещества А, cx – концентрация прореагировавшего вещества А за истекший промежуток времени; cA = cA0 − cx – концен-
трация вещества А в данный момент времени; α = cx 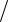 cA0 – степень превращения; k – константа скорости реакции.
cA0 – степень превращения; k – константа скорости реакции.
Из уравнения (2.5) следует, что размерность константы скорости для реакции первого порядка – обратное время [t–1]; для превращения данной части вещества требуется одинаковое время, независимо от исходной концентрации, т. е. в единицу времени превращению подвергнется одна и та же часть вещества. Примером реакций первого порядка служат радиоактивный распад, разложение простых эфиров при высоких температурах, процессы изомеризации.
Реакция второго порядка может быть представлена уравнением А + В → продукт (продукты) реакции
Её скорость в соответствии с уравнением (2.3) может быть выражена следующим уравнением:
− dc = kcAcB = k(cA0 |
− cx )(cB0 − cx ) |
(2.6) |
||||
dt |
|
|
|
|
||
или, после интегрирования: |
(cA0 |
−cx )cB0 |
|
|||
1 |
|
|
||||
k = |
|
ln |
|
|
(2.7) |
|
t(cA0 −cB0 ) |
(cB0 |
−cx )cA0 |
|
|||
36
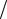
Если cA0 = cB0 , то выражение для константы скорости примет вид:
k = |
1 |
cx |
|
и cx = |
k (cA0 )2 t |
|
0 0 |
− cx ) |
0 |
||
|
t cA (cA |
|
1 + k (cA )t |
||
Если реакцию второго порядка схематически представить как 2A→ продукты,
то её скорость описывается дифференциальным уравнением
− dcdt = k(cA0 −2cx )2 .
Решение этого уравнения имеет следующий вид:
k = |
1 |
c |
x |
= |
c0 |
−c |
A |
|
||
|
|
|
|
A |
|
. |
||||
|
|
|
0 0 |
−2cx ) |
|
0 |
|
|||
|
|
t cA (cA |
|
2tcAcA |
||||||
(2.8)
(2.9)
(2.10)
Размерность константы скорости реакции второго порядка – [t–1c–1], поэтому, в отличие от константы скорости реакции первого порядка, числовое значение k зависит от того, в каких единицах выражены время t и концентрация с.
Если один из компонентов берётся в большом избытке, то его концентрация за время реакции практически не изменяется. Поэтому произведение его концентрации на константу скорости можно объединить в одну постоянную. При этом кинетика реакции формально описывается уравнением для реакций первого порядка с эффективной константой скорости k′. Значение константы скорости реакции второго порядка может быть определено делением эффективной константы скорости на концентрацию компонента, взятого в избытке. В этом случае молекулярность реакции, определяемая её механизмом, и порядок реакции, определяемый кинетическим уравнением, не будут совпадать.
Зависимость скорости реакции от температуры. С повышением температуры скорости реакций, как правило, увеличиваются: скорости большинства реакций возрастают в 2– 4 раза на каждые 10 К повышения температуры (правило Вант-Гоффа). Более точно температурная зависимость константы скорости описывается уравнением Аррениуса
k = B exp(−Ea RT ) , |
(2.11) |
в котором В – предэкспоненциальный множитель, почти не зависящий от температуры, Еа – энергия активации, величину которой можно определить из тангенса угла наклона прямой, уравнение которой получается логарифмированием уравнения Аррениуса:
ln k = ln B − |
Ea |
|
1 |
. |
(2.12) |
|
|
||||
|
R |
T |
|
||
37
Следовательно, для экспериментального определения величины энергии активации необходимо измерить константы скорости реакции при нескольких температурах и построить графическую зависимость ln k = f (1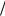 T ) . Энергия активации, обычно выражаемая в кДж/моль, – это избыток энергии по сравнению со средней энергией молекул при данной температуре, которым должны обладать молекулы, чтобы вступать в химическую реакцию.
T ) . Энергия активации, обычно выражаемая в кДж/моль, – это избыток энергии по сравнению со средней энергией молекул при данной температуре, которым должны обладать молекулы, чтобы вступать в химическую реакцию.
Экспериментальные методы определения скорости и порядка реакции
Измерение скорости реакции основано на определении концентрации одного из реагирующих веществ через различные промежутки времени от начала реакции. Для определения концентраций можно применять методы физико-химического анализа, основанные на зависимости физических свойств смеси от её состава (например, определение показателя преломления, угла вращения плоскости поляризации, вязкости, электрической проводимости, объёма, плотности, изменения температур замерзания и кипения, интенсивности окраски и т. п.), и методы аналитической химии (например, титрование). Поскольку концентрации по ходу реакции непрерывно меняются, то необходимо или очень быстрое измерение концентрации (методы физико-химического анализа), или торможение реакции во взятой пробе (химический контроль). Торможение может быть достигнуто охлаждением, резким разбавлением, устранением катализатора или совместным действием всех указанных факторов. Если реакция, протекающая в газовой фазе, сопровождается изменением числа молекул, то её течение удобно контролировать по изменению давления смеси во времени.
К сравнительно медленным реакциям со временем полупревращения порядка получаса и более можно применять спектроскопию, массспектрометрию и хроматографию. Для исследования скоростей очень быстрых реакций (с периодом полупревращения до 10-7 и даже 10–9 с) используются специально разработанные методы и особая аппаратура.
Для определения порядка реакции необходимо иметь экспериментальные данные об изменении концентрации реагирующих веществ со временем. Если в реакции участвует несколько веществ, то пользуются
методом изолирования Оствальда.
Допустим, в реакцию вступают три вещества: А, В и D
ν1А + ν2В + ν3 D →продукты
Скоростьэтойреакцииможетбытьвыраженакинетическимуравнением
− dcdt = kcAa cBb cDd ,
где a, b, c – порядки реакции по соответствующим компонентам.
Сначала для определения а проводят реакцию с большими избытка-
38
ми веществ В и D (концентрация вещества А равняется от 0,1 до 0,001 начальной концентрации всех остальных веществ). Тогда
− dcdt = k1caA ,
где k1=k cBb cDd . Затем проводят второй опыт с бóльшим избытком веществ А и В для определения d. В третьем опыте берут большой избыток А и D и определяют значение b. Таким образом, порядок реакции каждый раз искусственно снижается и сводится к определению частных порядков a, b и d. Общий порядок реакции равен сумме a+b+d. Все методы определения частных порядков можно разделить на две группы – интегральные и дифференциальные.
Интегральные методы. Здесь используются кинетические уравнения для определения скорости реакции в интегральной форме (полученные после интегрирования дифференциального уравнения скорости реакции). Разновидности этой группы методов.
1) Метод подбора уравнений, основанный на подстановке экспериментальных данных по концентрации вещества для каждого момента времени в кинетические уравнения реакций различных порядков. Определяемый порядок реакции соответствует тому уравнению, для которого при различных начальных концентрациях исходных веществ и в различные моменты времени при заданной температуре константа скорости будет оставаться постоянной.
1/c |
|
2 |
|
1/c=f(t) |
|
1/c2 |
|
|
|
|
|
ln(c) |
3 |
|
|
||
|
|
1/c2=f(t) |
|
|
|
ln(c)=f(t)
1
Время
Рис. 2.1. Зависимость различных функций концентрации исходных веществ от времени для реакций различных порядков.
2) При использовании графического метода определяют такую функциональную зависимость концентрации от времени, которая на графике даёт прямую линию (рис. 2.1). Для реакций первого порядка (пря-
39

мая 1) такой функцией является lnc; для реакций второго порядка (прямая 2) – 1/с ; для реакций третьего порядка (прямая 3) – 1/с2. Если концентрации исходных веществ различны, то для выделения частного порядка реакции по одному из компонентов аА, bB,,… используется метод изолирования Оствальда с последующим определением а и b. По тангенсу угла наклона полученной прямой вычисляют константу скорости реакции k;
3) Метод определения по периоду полупревращения τ1/2,
n =1+ ln(τ1′′/ 2 /τ1′/ 2 ) , ln(a′/ a′′)
где a и τ1/2 начальная концентрация и время полупревращения определенные для двух независимых экспериментов, проводимых в одинаковых условиях, но с различными начальными концентрациями исходных веществ. Те же соотношения сохраняются и при определении времени превращения любой доли исходной концентрации. Этот метод называют иногда методом Оствальда – Нойеса он позволяет определять произвольный порядок реакции, в том числе дробный и отрицательный.
Дифференциальные методы. Эти методы основаны на использовании для скорости реакции уравнения в дифференциальной форме.
По методу Вант-Гоффа реакцию проводят с компонентами, взятыми при двух различных исходных концентрациях a1 и a2. Тогда
n = ln(∆c / ∆t)1 − ln(∆c / ∆t)2 . ln a1 − ln a2
Графические варианты метода Вант-Гоффа основаны на использовании уравнения lnw= lnk + n lnc, которое получается при логарифмировании выражения для скорости реакции n-го порядка w=kcn . На графике в координатах lnw – lnс получается линейная зависимость с тангенсом угла наклона к оси ln с, соответствующим порядку реакции (tgϕ = n). Отрезок, отсекаемый на оси lnw, равен lnk. Скорость определяется тангенсом угла наклона касательной к кривой c = f(t) относительно оси времени. В зависимости от того, какая определяется скорость – в начальный момент реакции или в различные промежутки времени от начала реакции – различаются два графических варианта этого метода. Вариант 1 позволяет рассчитать концентрационный или истинный порядок nc реакции, так как скорость здесь определяется при отсутствии конечных или промежуточных продуктов, которые могут оказывать на неё влияние. В варианте 2 скорость определяется в различные моменты от начала реакции. Различие в величинах порядка реакции, определённого этими методами, позволяет обнаружить влияние на скорость реакции её конечных или промежуточных продуктов.
40