

IZBRANNYE_VOPROSY_KLINICHESKOI_GENETIKI
.pdfУниверсальных патологических повреждений соединительной ткани, которые бы формировали конкретный фенотип, не существует. Каждый дефект у каждого больного в своем роде уникален. При этом всеобъемлющее распространение в организме соединительной ткани определяет полиорганность поражений при ДСТ. В связи с этим предлагается подход с обособлением синдромов, связанных с диспластикозависимыми изменениями и патологическими состояниями.
Синдромы неврологических нарушений: вегетативная дисфункция (вегетососудистая дистония, панические атаки и др.), гемикрания. Вегетативная дисфункция формируется у значительного числа пациентов с ДСТ на раннем этапе— уже в раннем детском возрасте и рассматривается как обязательный компонент диспластического фенотипа. У большинства пациентов выявляется симпатикотония, реже встречается смешанная форма, в малом проценте случаев — ваготония. Выраженность клинических проявлений нарастает параллельно тяжести ДСТ. Вегетативная дисфункция отмечается в 97% случаев наследственных синдромов, при недифференцированной форме ДСТ — у 78% пациентов. В формировании вегетативных нарушений у пациентов с ДСТ, несомненно, имеют значение генетические факторы, лежащие в основе нарушения биохимизма обменных процессов в соединительной ткани и формировании морфологических субстратов, приводящих к изменению функции гипоталамуса, гипофиза, половых желез, симпатикоадреналовой системы.
Астенический синдром: снижение работоспособности, ухудшение переносимости физических и психоэмоциональных нагрузок, повышенная утомляемость. Астенический синдром выявляется в дошкольном и особенно ярко — в школьном, подростковом и молодом возрасте, сопровождая пациентов с ДСТ на протяжении всей жизни. Отмечается зависимость выраженности клинических проявлений астении от возраста больных: чем старше пациенты, тем больше субъективных жалоб.
Изменения клапанного аппарата сердца: изолированные и комбинированные пролапсы клапанов, миксоматозная дегенерация. Чаще изменения представлены пролапсом митрального клапана (ПМК) (до 70%), реже — пролапсами трикуспидального или аортального клапанов, расширением корня аорты и легочного ствола; аневризмами синусов Вальсальвы. В части случаев клапанная патология сопровождается явлениями регургитации, что отражается на показателях контрактильности миокарда и объемных параметрах сердца. Durlach J. (1994) предположил, что причиной ПМК при ДСТ может быть дефицит магния. Патология клапанов начинает формироваться также в детском возрасте (4–5 лет). Аускультативные признаки ПМК выявляются в различном возрасте: от 4 до 34 лет, однако наиболее часто — в возрасте 12–14 лет. Следует отметить, что эхокардиографические данные находятся в динамическом состоянии: более выраженные изменения отмечаются при последующих осмотрах, что отражает влияние возраста на состояние клапанного аппарата. Кроме того, на выраженность клапанных изменений влияет степень тяжести ДСТ и объем желудочков.
Торакодиафрагмальный синдром: астеническая форма грудной клетки, деформации грудной клетки (воронкообразная, килевидная), деформации позвоночника (сколиозы, кифосколиозы, гиперкифозы, гиперлордозы и др.), изменения стояния и экскурсии диафрагмы. Среди пациентов с ДСТ наиболее часто встречается воронкообразная деформация грудной клетки, на втором месте по частоте — килевидная деформация и наиболее редко выявляется астеническая форма грудной клетки. Начало формирование торакодиафрагмального синдрома приходится на ранний школьный возраст, отчетливость проявлений — на возраст 10–12 лет, максимальная выраженность — на период 14–15 лет. Наличие торакодиафрагмального синдрома определяет уменьшение дыхательной
поверхности легких, деформацию просвета трахеи и бронхов; смещение и ротацию сердца, «перекрут» основных сосудистых стволов. Качественные (вариант деформации) и количественные (степень деформации) характеристики торакодиафрагмального синдрома определяют характер и выраженность изменений морфофункциональных параметров сердца и легких. Деформации грудины, ребер, позвоночника и связанное с ними высокое стояние диафрагмы приводят к уменьшению грудной полости, повышению внутригрудного давления, нарушают приток и отток крови, способствуют возникновению аритмий сердца. Наличие торакодиафрагмального синдрома может повлечь за собой повышение давления в системе малого круга кровообращения.
«Торакодиафрагмальное сердце»: астенический, констриктивный, ложностенотический, псевдодилатационный варианты, торакодиафрагмальное легочное сердце. Формирование торакодиафрагмального сердца происходит параллельно манифестации и прогрессированию деформации грудной клетки и позвоночника, на фоне изменения клапанного аппарата и сосудов. Варианты торакодиафрагмального сердца служат отражением нарушения гармоничности взаимоотношений веса и объема сердца, веса и объема всего тела, объема сердца и объема больших артериальных стволов на фоне диспластикозависимой дезорганизации роста тканевых структур самого миокарда, в частности, его мышечных и нервных элементов.
Изменения сосудов: поражение артерий эластического типа: идиопатическое расширение стенки с формированием мешотчатой аневризмы; поражение артерий мышечного и смешанного типов: бифуркационно-гемодинамические аневризмы, долихоэктазии удлиненных и локальных расширений артерий, патологическая извитость вплоть до петлеобразования; поражение вен (патологическая извитость, варикозное расширение вен верхних и нижних конечностей, геморроидальных и др. вен); телеангиоэктазии; эндотелиальная дисфункция. Сосудистые изменения сопровождаются повышением тонуса в системе крупных, мелких артерий и артериол, уменьшением объема и скорости наполнения артериального русла, снижением венозного тонуса и избыточным депонированием крови в периферических венах и, как правило, манифестируют в подростковом и молодом возрасте, прогрессируя с увеличением возраста пациентов.
Изменения артериального давления: идиопатическая артериальная гипотензия.
Нарушение метаболизма миокарда: кардиалгии, аритмии сердца, нарушения процессов реполяризации (I степень: увеличение амплитуды зубца Т V2-V3, зубец Т V2 > Т V3; II степень: инверсия зубца Т, смещение ST V2-V3 вниз на 0,5–1,0 мм; III степень: инверсия зубца Т, косовосходящее смещение ST до 2,0 мм). Нарушение метаболизма миокарда определяется влиянием кардиальных факторов (изменения клапанного аппарата, варианты торакодиафрагмального сердца) и экстракардиальных условий (торакодиафрагмальный синдром, вегетативная дисфункция, сосудистые изменения, дефицит микро- и макроэлементов). Метаболические нарушения при ДСТ не имеют специфических субъективных симптомов и клинических проявлений, вместе с тем потенциально определяют повышенный риск внезапной смерти в молодом возрасте с преобладающей ролью в танатогенезе аритмий сердца.
Аритмии сердца: желудочковая экстрасистолия различных градаций; многофокусная, мономорфная, реже полиморфная, монофокусная предсердная экстрасистолия; пароксизмальные тахиаритмии; миграция водителя ритма; атриовентрикулярные и внутрижелудочковые блокады; аномалии проведения импульса по дополнительным путям; синдром предвозбуждения желудочков; синдром удлинения интервала QT. Частота выявления аритмии при ДСТ— около 64%. Источником нарушения ритма сердца может
быть очаг нарушенного метаболизма в миокарде. При нарушении структуры и функции соединительной ткани всегда присутствует подобный субстрат биохимического генеза. Причиной нарушений сердечного ритма при ДСТ могут служить клапанные изменения. Возникновение аритмий при этом может быть обусловлено сильным натяжением митральных створок, содержащих мышечные волокна, способные к диастолической деполяризации с формированием биоэлектрической нестабильности миокарда. Кроме того, появлению аритмий может способствовать резкий сброс крови в левый желудочек с пролонгированной диастолической деполяризацией. Изменения геометрии камер сердца также могут иметь значение в возникновении аритмий при формировании диспластического сердца, особенно торакодиафрагмального варианта легочного сердца. Кроме кардиальных причин происхождения аритмий при ДСТ существуют и экстракардиальные, обусловленные нарушением функционального состояния симпатического и блуждающего нервов, механического раздражения перикарда деформированным костяком грудной клетки. Одним из аритмогенных факторов может быть дефицит магния, выявляемый у пациентов с ДСТ. В исследованиях российских и зарубежных авторов получены убедительные данные о причинной взаимосвязи между желудочковыми и предсердными аритмиями и внутриклеточным содержанием магния. Предполагается, что гипомагниемия может способствовать развитию гипокалиемии. При этом увеличивается мембранный потенциал покоя, нарушаются процессы деполяризации и реполяризации, снижается возбудимость клетки. Замедляется проводимость электрического импульса, что способствует развитию аритмий. С другой стороны, внутриклеточный дефицит магния повышает активность синусового узла, снижает абсолютную и удлиняет относительную рефрактерность.
Внезапная сердечная смерть: изменения сердечно-сосудистой системы при ДСТ, определяющие патогенез внезапной смерти, — патологии клапанного аппарата, сосудов, аритмии сердца. По наблюдениям во всех случаях причина смерти непосредственно или опосредованно связана с морфофункциональными изменениями сердца и сосудов: в одних случаях она обусловлена грубой сосудистой патологией, которую легко констатировать на вскрытии (разрывы аневризм аорты, артерий головного мозга и др.), в других случаях внезапная смерть вызвана факторами, трудно поддающимися верификации на секционном столе (аритмическая смерть).
Патология системы внешнего дыхания: трахеобронхиальная дискинезия, трахеобронхомаляция, трахеобронхомегалия, вентиляционные нарушения (обструктивные, рестриктивные, смешанные нарушения), спонтанный пневмоторакс. Бронхолегочные нарушения при ДСТ современные авторы описывают как генетически обусловленные нарушения архитектоники легочной ткани в виде деструкции межальвеолярных перегородок и недоразвития эластических и мышечных волокон в мелких бронхах и бронхиолах, ведущие к повышенной растяжимости и сниженной эластичности легочной ткани.
Изменение функциональных параметров дыхательной системы при ДСТ зависит от наличия и степени деформации грудной клетки, позвоночника и чаще характеризуется рестриктивным типом вентиляционных нарушений со снижением общей емкости легких (ОЕЛ). Остаточный объем легких (ООЛ) у многих пациентов с ДСТ не меняется или слегка повышается без изменения соотношения объема форсированного выдоха в первую секунду (ОФВ1) и форсированной жизненной емкости легких (ФЖЕЛ). У некоторых пациентов выявляются обструктивные нарушения, феномен гиперреактивности бронхов, что пока не нашло однозначного объяснения. Пациенты с ДСТ представляют собой группу с высоким риском возникновения ассоциированной патологии, в частности, туберкулеза легких.
Иммунологические нарушения: иммунодефицит, аутоиммунный синдром, аллергические реакции. Функциональное состояние иммунной системы при ДСТ характеризуется как активацией иммунных механизмов, обеспечивающих поддержание гомеостаза, так и их недостаточностью, ведущей к нарушению способности адекватно освобождать организм от чужеродных частиц и, следовательно, к развитию рецидивирующих инфекционновоспалительных заболеваний бронхолегочной системы, у части пациентов с ДСТ включают повышение в крови уровня иммуноглобулина Е. В целом, литературные данные о сдвигах в иммунной системе при различных клинических вариантах ДСТ носят неоднозначный, нередко противоречивый характер, что требует дальнейшего их изучения, а механизмы их формирования при ДСТ до сих пор остаются практически неизученными.
Нарушение положения, структуры и функции внутренних органов: нефроптоз и дистопии почек, птозы органов желудочно-кишечного тракта, органов малого таза, дискинезии органов желудочно-кишечного тракта, дуоденогастральные и гастроэзофагеальные рефлюксы, несостоятельность сфинктеров, дивертикулы пищевода, грыжи пищеводного отверстия диафрагмы; птозы половых органов у женщин.
Патология органа зрения: миопия, астигматизм, гиперметропия, косоглазие, нистагм, отслойка сетчатки, вывих и подвывих хрусталика. Нарушения аккомодации проявляется в различные периоды жизни, у большинства обследованных — в школьные годы (8–15 лет) и прогрессирует до 20–25 лет.
Геморрагические гематомезенхимальные дисплазии: гемоглобинопатии, синдром Рандю– Ослера–Вебера, рецидивирующие геморрагические (наследственная дисфункция тромбоцитов, синдром Виллебранда, комбинированные варианты) и тромботические (гиперагрегация тромбоцитов, первичный антифосфолипидный синдром, гипергомоцистеинемия, резистентность фактора Vа к активированному протеину С) синдромы.
Патология стопы: косолапость, плоскостопие (продольное, поперечное), полая стопа. Эти проявления ДСТ являются одним из самым ранних проявлений несостоятельности соединительнотканных структур. Наиболее часто встречается поперечно-распластанная стопа (поперечное плоскостопие), в части случаев сочетающаяся с отклонением 1 пальца наружу (hallus valgus) и продольное плоскостопие с пронацией стопы (плосковальгусная стопа). Патологические изменения стопы еще больше уменьшает возможность физического развития пациентов с ДСТ, формирует определенный стереотип жизни, усугубляет психосоциальные проблемы.
Синдром гипермобильности суставов: нестабильность суставов, вывихи и подвывихи суставов. Синдром гипермобильности суставов в большинстве случаев определяется уже в раннем детском возрасте. Максимальная гипермобильность суставов наблюдается в возрасте 13–14 лет, к 25–30 годам распространенность снижается в 3–5 раз. Частота встречаемости гипермобильности суставов достоверно выше среди пациентов с выраженной ДСТ.
Изменения позвоночника: ювенильный остеохондроз позвоночника, нестабильность, межпозвонковые грыжи, вертебробазиллярная недостаточность; спондилолистез. Развиваясь параллельно развитию торакодиафрагмального синдрома и синдрома гипермобильности, эта группа патологии существенно усугубляет их последствия.
Диспластикозависимые косметические изменения: дисморфии челюстно-лицевой области (аномалии прикуса, готическое небо, выраженные асимметрии лица); О- и Х-образные
деформации конечностей; изменения кожных покровов (тонкая просвечивающаяся и легко ранимая кожа, повышенная растяжимость кожи, шов в виде «папиросной бумаги»). Данные проявления ДСТ значительно усугубляются наличием малых аномалий развития, выявляемых у абсолютного большинства пациентов с ДСТ. При этом подавляющее большинство пациентов имеет 1–5 микроаномалий (гипертелоризм, гипотелоризм, «мятые» ушные раковины, большие торчащие уши, низкий рост волос на лбу и шее, кривошея, диастема, неправильный рост зубов и др.).
Нарушения психической сферы: невротические расстройства, депрессии, тревожность, ипохондрия, обсессивно-фобические расстройства, нервная анорексия. Известно, что пациенты с ДСТ формируют группу повышенного психологического риска, характеризующуюся сниженной субъективной оценкой собственных возможностей, уровнем претензий, эмоциональной устойчивости и работоспособности, повышенным уровнем тревожности, ранимостью, депрессивностью, конформизмом. Наличие диспластикозависимых косметических изменений в сочетании с астенией формируют психологические особенности этих больных: сниженное настроение, потеря ощущения удовольствия и интереса к деятельности, эмоциональная лабильность, пессимистическая оценка будущего, нередко с идеями самобичевания и суицидальными мыслями. Закономерным следствием психологического дистресса является ограничение социальной активности, ухудшение качества жизни и значительное снижение социальной адаптации, наиболее актуальные в подростковом и молодом возрасте.
Малые аномалии развития
MAP - изменения структуры различных органов и тканей, не сопровождающиеся клинически значимыми нарушениями их функции. MAP следует разделять на внешние и висцеральные. К внешним относятся аномалии развития кожи и костей черепа, кисти и стопы: гиперпигментация и депигментация кожи, оттопыренные уши, отсутствие мочки уха, синдактилия, сандалиевидная щель и др. К висцеральным - изменения строения внутренних органов: удвоение чашечно-лоханочного аппарата почек, добавочная доля селезенки и др., а также перечисленные выше MAC. У большинства здоровых лиц удается обнаружить до 3-4 MAP. Среднее количество MAP у лиц с ДСТ достоверно выше, чем в популяции, что, возможно, подтверждает их патогенетическую связь.
MAP обнаруживают при рождении или в раннем детском возрасте, часть из них поддается обратному развитию: ООО, увеличенный евстахиев клапан и пр. Другие сохраняются в течение жизни, однако с возрастом могут приобретать самостоятельное клиническое значение, способствуя развитию патологии или становясь фактором риска (ФР) кардиальной патологии.
На основе результатов фенотипического исследования, изучения семьи, анализа внешних и висцеральных признаков врач вправе заподозрить наличие ННСТ. Все это делает необходимым освещение вопросов диагностики наиболее распространенных моногенных ННСТ.
Синдром Марфана
СМ - аутосомно-доминантное, мультисистемное HHCT, характеризующееся высокой вариабельностью клинических проявлений. Его диагностика сегодня по-прежнему основана на Гентских критериях, пересматриваемых в настоящее время. В основе алгоритма диагностики СМ лежит определение больших и малых критериев, характеризующих степень выраженности изменений СТ в различных органах и системах (таблица 1). При наличии у пациента "Гентских критериев" СМ рекомендуется провести молекулярно-генетическое исследование с целью поиска мутаций генов, кодирующих фибриллин. Часть перечисленных синдромов при

отсутствии полного набора 'Тентских критериев" СМ и при возможности молекулярногенетического обследования неизбежно окажется включенной в MПФ. Принципы диагностики МПФ будут изложены далее.
Таблица 1 «Гентские критерии» диагностики синдрома Марфана
Большие критерии |
Малые критерии |
Костные |
|
Наличие 4 критериев из 8 следующих: |
- Умеренная воронкообразная деформация грудной |
- Килевидная деформация грудной клетки, |
клетки |
требующая хирургического вмешательства |
- ГМС |
- Отношение верхнего сегмента тела к нижнему < |
- Арковидное небо со скученностью зубов |
0,86 или отношение между размахмом рук и ростом |
- Деформация черепа: долихоцефалия, гипоплазия |
≥ 1,05 |
скуловых костей, энофтальм, скошенные глазные |
- Положительный тест запястья и большого пальца |
щели, ретрогнатия |
(Штейнберг) |
|
- Сколиоз › 20° или спондилолистез - Контрактура локтевого сустава с ограничением
выпрямления локтевого сустава < 170° - Медиальное смещение внутренней лодыжки, приводящее к плоскостопию
- Протрузия вертлужной впадины любой степени, подтвержденная Rg
Изменения в костной системе соответствуют большому критерию, - патологически значимые изменения, если обнаружены не менее 4 из перечисленных 8 больших критериев Костная система вовлечена, если обнаружены не менее 2 больших критериев или 1 большой и 2 малых
Глазные
Подвывих хрусталика |
Аномально плоская роговица (по результатам |
|
кератометрических измерений); |
|
Удлинение переднезадней оси глазного яблока (по |
|
данным УЗИ) с миопией |
|
Гипоплазия радужной оболочки и/или цилиарной |
|
мышцы с затруднением миоза |
Зрительная система вовлечена, если определены 2 малых критерия |
|
Сердечно-сосудистая система |
|
Расширение восходящей аорты с аортальной |
ПМК |
регургитацией или без неё и вовлечением, как |
Расширение ствола легочной артерии при отсутствии |
минимум, синусов Вальсальвы; или |
клапанного или периферического легочного стеноза |
Расслоение восходящей аорты |
или любой другой очевидной причины в возрасте < |
|
40 лет |
Обызвествление митрального кольца в возрасте < 40 лет Расширение/расслоение стенки грудной или
брюшной аорты в возрасте < 50 лет Сердечно-сосудистая система вовлечена, если выявлен 1 большой или 1 малый критерий
|
Бронхо-легочная система |
Отсутствуют |
Спонтанный пневмоторакс |
|
Апикальные буллы, подтвержденные Rg грудной |
|
клетки |
Бронхо-легочная система вовлечена, если выявлен 1 малый критерий
Кожные
Отсутствуют Атрофические стрии, не связанные с выраженными изменениями массы тела, беременностью или частым локальным механическим воздействием Рецидивирующие или послеоперационные грыжи
Кожа вовлечена, если выявлен 1 малый критерий
Твердая мозговая оболочка
Пояснично-крестцовая дуральная эктазия, |
Отсутствуют |
выявленная при КТ или МРТ |
|
Отягощенная наследственность |
|
Наличие близких родственников со следующими |
Отсутствуют |
диагностическими критериями: Мутация в FBN1, известная как причина
возникновения СМ; или выявление ДНК-маркеров СМ  Вовлечение при наличии 1 большого критерия
Вовлечение при наличии 1 большого критерия
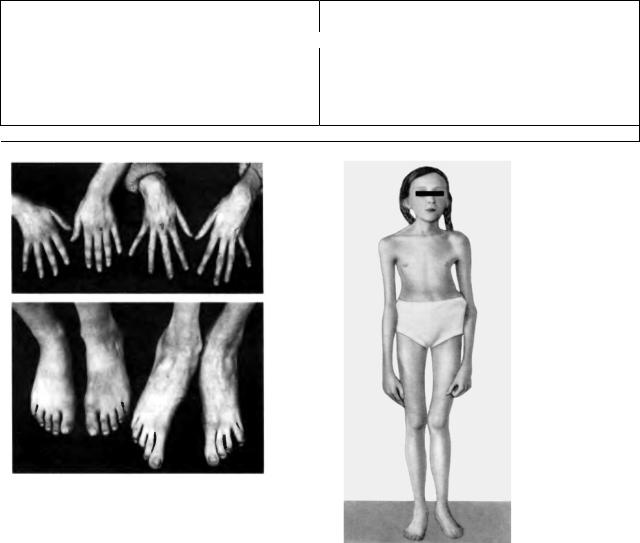
Рис.1: Кисти и стопы в норме (слева) и при синдроме Марфана (справа). Рис.2: Пример женского фенотипа при синдроме Марфана
Синдром Элерса-Данло
Диагностика СЭД в настоящее время основана на "Вилльфраншских критериях". В них вместо ранее признанных 10 выделены 6 типов: классический, гипермобильный, сосудистый, кифосколиотический, артрохалазия, дерматоспараксис. Большие и малые диагностические критерии определены для каждого типа и дополнены по мере возможности результатами лабораторных исследований.
В таблице 2 представлена классификация наиболее распространенных типов СЭД. Для клинической диагностики необходимо наличие хотя бы 1 большого критерия. Малые критерии имеют более низкий уровень диагностической специфичности. Наличие ≥ 1 малых критериев вносит вклад в диагностику того или иного типа СЭД. При отсутствии больших критериев малые недостаточны для диагностики. Наличие малых критериев дает основание подозревать состояние, подобное СЭД, характер которого будет ясен по мере того, как станет известной его молекулярная основа.
Помимо СЭД, перечисленные признаки ННСТ могут соответствовать еще целому ряду наследственных заболеваний (семейная ГМС (Joint laxity, familial), синдром вялой кожи (Cutis laxa), синдром затылочного рога (Occipital Horn syndrome)). Наряду с наличием СЭД, полностью отвечающего "Вилльфраншским критериям", во многих случаях имеется их неполный набор. Такие случаи следует относить к элерсоподобному фенотипу (ЭПФ), критерии диагностики которого будут изложены далее.
Таблица 2 «Вильфраншские критерии» диагностики синдрома Элерса-Данло
Большие критерии |
Малые критерии |
Молекулярный |
|
Классический тип, АДт |
дефект |
|
|
|
Повышенная |
Гладкая, бархатистая кожа с повышенным ростом |
Pro alfa 1 (V) или |
растяжимость кожи. |
пушковых волос. |
Pro alfa 2 (V) |
Широкие |
Моллюсковидные псевдоопухоли. |
коллаген цепей типа |
атрофические рубцы |
Подкожные сферические образования. |
V. |
|
Осложнения ГМС: растяжение сустава, вывихи и |
Ненормальная |
|
подвывихи, плоскостопие. |
структура волокон |
|
Мышечная гипотония, задержка развития моторики. |
коллагена по типу |
|
Легкое образование гематом при незначительных травмах. |
«цветной капусты». |
|
Выраженные проявления растяжимости и слабости тканей |
|
|
(грыжа пищеводного отверстия, анальный пролапс в |
|
|
детском возрасте, цервикальная недостаточность). |
|
|
Послеоперационные грыжи. |
|
|
Наличие аналогичных заболеваний в семье. |
|
|
Гипермобильный тип, АДт |
|
Кожные |
Рецидивирующие смещения (подвывихи) суставов. |
Мутации генов |
патологические |
Хронические боли в суставах/конечностях. |
синтеза коллагена |
проявления |
Наличие аналогичных заболеваний в семье. |
III L1, тенасцина Х. |
(гиперрастяжимость |
|
|
и/или гладкая, |
|
|
бархатистая кожа). |
|
|
Генерализованная |
|
|
ГМС. |
Сосудистый тип АДт |
|
|
|
|
Тонкая, |
Акрогерия. |
Аномальная |
просвечивающая кожа. |
Гипермобильность малых суставов. |
структура коллагена |
Слабость или разрывы |
Разрыв сухожилий и мышц. |
III, |
артерий, кишечника, |
Эквиноварусная деформация стопы (косолапость). |
вырабатываемого |
матки. |
Варикозные вены в юношеском возрасте. |
фибробластами; |
Обширные |
Артериовенозные каротидно-кавернозные фистулы. |
мутации гена |
кровоподтеки и |
Пневмоторакс/пневмогемоторакс. |
COL3A1 |
поверхностные |
Рецессия десны. |
|
травмы. |
Наличие аналогичных заболеваний в семье. |
|
Характерный вид лица. |
ВС близких родственников. |
|
|
Кифосколиотический тип АРт |
|
Генерализованная |
Легкая ранимость кожи, атрофические рубцы. |
PLOD1, |
ГМС. |
Наклонность к гематомам |
лизил-4- |
Тяжелая мышечная |
Разрывы артерий. |
гидроксилаза 1 |
гипотония с рождения. |
MB. |
|
Прогрессирующий |
Уменьшение размеров роговицы |
|
самопроизвольная |
Rg значимое нарушение остеогенеза. |
|
отслойка сетчатки, |
Семейный анамнез, например, болезнь сибсов. |
|
миопия, глаукома |
Артрохалазия АДт |
|
|
|
|
Тяжелая |
Повышенная растяжимость кожи. |
COL1A1, COL1A2 |
генерализованная ГМС |
Ранимость кожи, атрофические рубцы. |
коллаген I типа |
с повторными |
Легко возникающие гематомы |
|
подвывихами. |
Мышечная гипотония |
|
Врожденная |
Кифосколиоз |
|
дислокация |
Легкий остеопороз (радиологическое исследование) |
|
тазобедренных и |
|
|
других крупных |
|
|
суставов. |
|
|
Дерматоспараксис АРт (Х-сцепленное рецессивное заболевание) Недостаточная активность проколлаген-пептидазы
Тяжелая форма |
Мягкая, рыхлая структура кожи. |
ADAMTS2 |
слабости кожи. |
Легко возникающие гематомы. |
проколагеновая N- |
Провисающая, |
Преждевременный разрыв плодных оболочек. |
протеиназа |
излишняя кожа. |
Большие грыжи (пуповинные, паховые). |
|
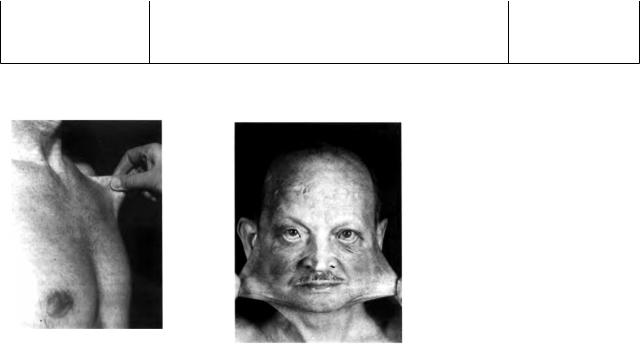
Рис.3: Повышенная растяжимость кожи под ключицей (слева), кожи лица, рубцы на лбу (справа)
Несовершенный остеогенез
НО (osteogenesis imperfecta) - группа ННСТ, характеризующаяся повышенной ломкостью костей. Для его диагностики до настоящего времени используют маркеры. НО относится к заболеваниям АДт наследования, однако возможны спонтанные мутации. Выделяют 8 типов НО, для которых характерны или недостаточное количество, или низкое качество коллагена. Основными клиническими признаками являются повышенные ломкость и деформация костей, слабость связочного аппарата суставов, низкий мышечный тонус, малый рост и голубые склеры.
Синдром гипермобильности суставов
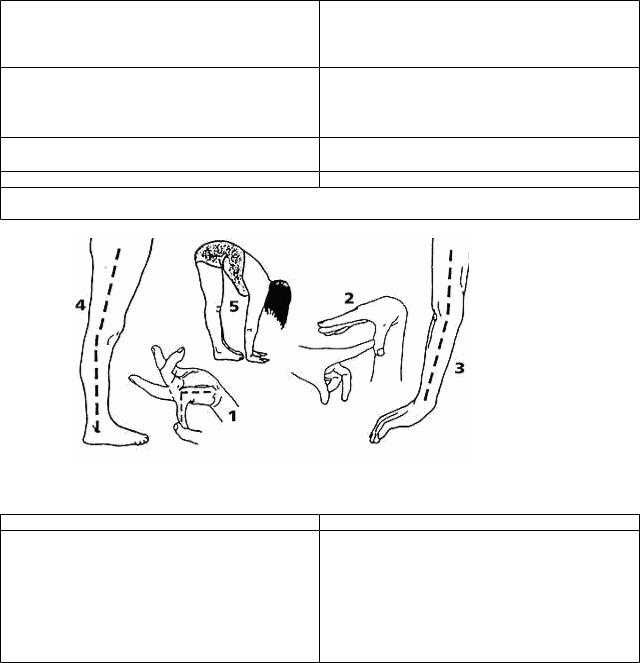
Из всех ННСТ с согласованными критериями клинической диагностики, СГМС наиболее распространен в клинической практике. Гипермобильными следует считать суставы с избыточным объемом движений. Оценивая ГМС, следует учитывать возраст, пол и этническое происхождение пациента. Известно, что у здоровых людей суставная мобильность снижается с возрастом, у женщин ее уровень выше, чем у мужчин, у выходцев из Азии она наибольшая, у европейцев наименьшая. Выраженность ГМС следует оценивать по девятибальной шкале Beighton Р (таблица 3).
ГМС является результатом слабости связок, которая носит наследственными характер. Особую роль в этом отношении играют мутации генов, кодирующих коллаген, эластин, фибриллин и тенасцин X.
ГМС может быть приобретённой, поскольку объем движений в суставах может быть увеличен до гипермобильного под воздействием тренировок. Артистам балета, не обладающим наследственной повышенной растяжимостью связок, приходится развивать гипермобильность определенных суставов, при этом изначально неизмененные околосуставные ткани защищают их от травм.
СГМС следует называть сочетание признаков ГМС с клинической симптоматикой. Речь идет о частых вывихах и подвывихах суставов, артралгиях, вовлечении ВНС (ВД - вегетативная дисфункция). Таким образом, для понимания взаимосвязей между ГМС и СГМС уместно привести формулу Grahame R: ГМС + симптоматика = СГМС.
Клинические признаки СГМС частично совпадают с таковыми при других ННСТ. К ним, помимо ГМС, относятся повышенная растяжимость кожи, нарушение рубцевания
и стрии, марфаноидная внешность (MB), а также остеопения. СГМС, хотя и не уменьшает продолжительности жизни, существенно снижает ее качество, т. к. сопровождается возникновением суставных болей и нетрудоспособностью.
В таблице 4 приведены критерии СГМС.
СГМС диагностируют при наличии 2 больших критериев либо 1 большого и 2 малых, или 4 малых критериев. 2 малых критерия достаточно, если имеется близкий родственник, страдающий данным заболеванием. СГМС исключают при наличии СМ или СЭД, иных типов, кроме гипермобильного типа ЭДС, в соответствии с определениями, предусмотренными "Гентскими" и "Вилльфраншскими критериями".
Таким образом, диагноз СГМС устанавливают при наличии ГМС и суставных болей, предварительно исключив СМ и СЭД. Диагноз СГМС у части пациентов в настоящее время может быть подтвержден лабораторным анализом уровня тенасцина X сыворотки крови и при определении полиморфизма гена тенасцина X.
Таблица 3 |
Девятибалльная шкала ГМС |
|
||
|
Тест |
|
|
Суставы |
1. |
Способность |
правый |
левый |
|
Пассивно разогнуть V палец в |
1 |
1 |
||
2. |
пястнофаланговом суставе > 90° |
1 |
1 |
|
Пассивно привести 1 палец к ладонной |
||||
3. |
поверхности руки |
|
1 |
1 |
Пассивно разогнуть локтевой сустав ≥ 10° |
||||
4. |
Пассивно разогнуть коленный сустав ≥ 10° |
1 |
1 |
|
5. |
Интенсивно прижать ладони к полу, не |
|
1 |
|
Итого |
сгибая коленей |
|
|
9 |
|
|
|
||
Один балл может быть получен для каждой стороны при манипуляциях 1-4, поэтому показатель ГМС максимально составляет 9 баллов
Таблица 4 Пересмотренные диагностические критерии СГМС
Большие критерии |
Малые критерии |
Показатель Beighton 4/9 или выше (как при |
Показатель Beighton 1,2 или 3/9 (0,1,2 или 3, |
обследовании, так и в прошлом)* |
у пациента ≥ 50 лет)*. |
Артралгия > 4 суставов в течение > 3 |
Артралгия >3 месяцев в 1-3 суставов или |
месяцев * |
боль в спине > 3 мес.), спондилез, |
|
спондилез/спондилолистез*. |
|
Смещение/подвывих > 1 сустава или 1 |
|
сустава с неоднократным повторением. |
|
Воспаление мягких околосуставных тканей. |