

Глава 2.
ПОВЕДЕНИЕ БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ В ВОДНОЙ СРЕДЕ
В настоящей главе приведены результаты исследований и их обсуждение по всем работам автора, касающимся всевохможных аспектов состояния ряда биологически активных соединений, в основном, производных аденозинфосфатов в водной среде. Получение данных о превращениях на молекулярном уровне производилось методом протонного магнитного резонанса (ПМР), что в немалой степени способствовало полному пониманию происходящих процессов, т.е. до расшифровки элементарных стадий.
К специфике предлагаемого изложения следует отнести выделение биологического аспекта исследований, оставляя строгие физико-химические доказательства с необходимыми математическими выкладками, а также исходный эмпирический материал цитируемым опубликованным статьям автора. На наш взгляд это позволяет сохранить целостность и ясность представляемой работы.
В последовательности превращений изучаемых соединений в водной среде удалось получить интересные данные о конформационных переходах, полиассоциации, внутримолекулярном свёртывании,протонизации и межмолекулярном взаимодействии, что позволило сформулировать общие принципы образования вторичных структур в водной среде.
2-1. Конформационный переход“син-антимв аденозин-б^- фосфате. При изучении поворота аденинового основания вокруг С-Nсвязи[55] была получена величина энергетического барьера,
39
составившая 2 ккал/моль,что оказалось близким к расчётным значениям [91,146]. В положении“анти”, т.е. когда основание как бы отвёрнуто от рибозы, находится при комнатной температуре 2/3 молекул.
Получение количественных характеристик перехода “син-анти”позволяет утверждать, что в молекуле аденозин-5-фосфата происходит достаточно свободное вращение основания вокругC-Nсвязи. Вследствие того,что энергия активации перехода“анти-син” близка по величине к энергии теплового движения, разница в энергетических уровнях син и анти форм проявится только при уменьшении количества свободно сталкивающихся с АМФ молекул. Это может быть в том случае, если нуклеотид окажется стерически частично или полностью закрыт(напрмер, в случае образования ассоциатов или полинуклеотидной цепи). Поэтому следует ожидать преимущественного анти расположения аденинового основания в более сложных образованиях, включающих аденозин-5;-фосфат и создающих внешние стерические затруднения для вращения по гликозидной связи. (Заместители в самой молекуле аденозин-5у-фосфата очевидно могут стабилизировать как син, так и анти положения).
О роли водной среды в переходе “син-анти”может свидетельствовать лишь величина изменения энтропии -5,4 э.е. [55]. Для конформационного перехода это значимая величина, которая указывает на относительную лёгкость раздвижения молекул растворителя при переходе. Скорее всего в жёсткой сетке водородных связей следовало бы ожидать значительно большего изменения энтропии.
40
2-2. Пол иассоциацияаденошн-5-фосфатов. Идея исследования отдельных стадий ассоциации может быть осуществлена при выполнении ряда условий. В зависимости от величин констант ассоциации в определённых областях концентрации будет преобладать влияние какого-либо одного ассоциата. Чем более разбавленный раствор, тем меньшее число ассоциатов вносит свой вклад в экспериментальную величину измеряемого химического сдвига и тем легче разделить эти вклады. С увеличением концентрации, и соответственно, числа различных ассоциатов изменения в ходе кривой, связанные с преимущественным влиянием какой-либо стадии, должны уменьшаться и с определённого значенияч концентрации не будут превышать ошибки измерения. Вследствие этого при больших концентрациях наблюдаемая зависимость не может дать какие-либо сведения о механизме полиассоциации. Таким образом, следует производить измерения только в области малых концентраций, в которой только начинает проявляться влияние ассоциатов. В эксперименте это обычно отмечается как начало отклонения от линейной зависимости измеряемого параметра от исходной концентрации. Необходимо отметить также, что для выявления точного хода кривой требуется достаточное количество экспериментальных точек. При условии выполнения данных требований нами предложена схема учёта влияния всевозможных ассоциатов на примере изучения состояния аденозинмонофосфатов [27]. Нами показано, что вначале происходит образование стопки оснований с величиной константы ассоциации в стадии образования димеров К=11,5 л/моль. Однако модель последовательного присоединения оснований для увеличения стопки не соответствовала экспериментальным данным и после стадии димеризации пришлось предпола-
41
гать взаимопроникновение димеров, которое происходит легче, чем упаковка оснований в стопку. Интересно, что такие взаимопроникающие димеры в чём-то напоминают модель элемента двухспиральных нуклеиновых кислот и это позволяет предполагать соответствующий возможный механизм построения полинуклеотидных цепей.
Таким образом, естественное,казалось бы, предположение о стопкообразовании нуклеотидов, которое вытекает из идеи максимального перекрывания гетероциклов, в результате полученных данных о ступенчатых константах ассоциации [27] претерпело существенное изменение. Если бы образование стопки происходило только за счёт химического сродства адениновых колец, то в эксперименте наблюдалось бы увеличение числа единиц в стопке. Однако, после тщательного физико-химического анализа оказалось предпочтительнее образование из димеров не стопки, чуть ли не фрагмента двойной спирали. В этом наглядна проявилась определяющая роль водной среды, спокойно изменяющей механизм ассоциации вследствие его гидрофобного характера, т.е. при появлении большей площади соприкосновения, чем один гетероцикл, гидрофобный процесс предпочитает этот новый более выгодный для ассоциации путь.
Прежде чем более подробно излагать экспериментальные данные, подтверждающие гиброфобную природу взаимодействия нуклеотидов, необходимо было исследовать влияние на структуру ассоциатов появления зарядов на адениновом основании и фосфатной группе.
РОССИЙСКАЯ
гсзсу^.'.^'пу-м*
42
2-3. Исследование протон изацни ад енозин-5-фосфатов [37]
позволило,с одной стороны,подтвердить конформацию“син-анти”при нейтральных pH (6,5), с другой стороны полученные величины изменений энтальпии(0ккал/моль) и энтропии(31э.е.) при ионизации фосфатной группы свидетельствуют о сопутствующем пере- структурировании водной среды, окружающей аденозин-5/- ффосфат. Этого вывода вполне достаточно для более тщательного изучения гидрофобного характера взаимодействия нуклеотидов.
2-4. Исследование природы межмолекулярных взаимодействий аденозин-^-фосфатов.Наиболее информативной, хотя и непривычной по виду, оказалась полученная зависимость [27] логарифма контанты димеризации аденозин-5/-фосфата от обратной температуры (рис.З). Указанная зависимость оказывается нелинейной и позволяет оценить только интервалы изменений энтальпии(
0,3 - 12ккал/моль)и энтропии(7-44 э.е.) в выбранном диапазоне температур. Как видно из рис.З с увеличением температуры константа димеризации растёт, а величины изменений энтальпии и энтропии падают. Данный ход зависимости свидетельствует о том, что состояние воды, характерное для каждого значения температуры,является определяющим при ассоциации АМФ.
Из приведённых фактов вытекает лишь одно представление о структуре ассоциатов в указанной области температур, а именно, то, что расстояние между основаниями в ассоциате должно быть максимальным. Вероятно, это зависит от механизма взаимодействия АМФ с молекулами воды, что, в свою очередь, во многом определяется структурой самой воды. Определяющее влияние среды на образование димеров и тетрамеров свидетельствует о гидрофобной природе взаимодействия. Мы не ставили перед собой задачу выяс
43
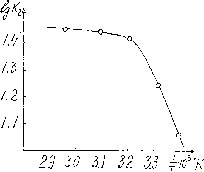
Рис.3Зависимость логарифма константы димеризации аде- нозин - 5''- фосфата от обратной температуры.
44
нения точной структуры димера, данные о которой уже приведены в некоторых работах [118,192,197]. Кроме того, нам представляется, что в растворе всегда имеется набор различных структур димеров, находящихся в динамическом равновесии, и речь может идти только об эффективной структуре. По нашему мнению, более важным является понимание природы последовательного образования ассоциатов. Для выяснения природы взаимодействия между молекулами АМФ мы использовали данные о тетрамере [28].Необходимо иметь ввиду, что мономеры, димеры, тетрамеры и последующие ассоциаты постоянно обмениваются между собой молеулами АМФ.
По нашему мнению,наиболее устойчивым состоянием должно быть состояние тетрамера в виде “четырёхугольной звезды”, поскольку вероятность взаимодействия гетероцикла с каждым из двух соседних оснований примерно одинакова.
Таким образом, совокупность полученных данных говорит о гидрофобном характере взаимодействия гетероциклов и позволяет представить механизм образования первичных ассоциатов АМФ в воде.
2-5. Некоторые выводы об образовании ассоциатов АМФ в воде. Анализ литературных данных (гл.1) и полученных экспериментальных результатов (гл.2) позволяет произвести дополн/итель- ные рассуждения о природе образования ассоциатов АМФ. На наш взгляд, есть основания охарактеризовать поведение молекул АМФ в воде следующим образом. Во-первых, процесс растворения данных нуклеотидов в воде приводит к их гидратации в местах, яв
45
ляющихся центрами образования водородной связи (рис.2). Следует учитывать, что рисунок соответствует состоянию АМФ в воде при значениях pH, близких к нейтральному. В этом случае депро- тонированатолько одна группа ОН в фосфате. Во-вторых, атака молекул Н20 на центры образования водородной связи приведёт к обмену протонов воды с протонами гидроксильных групп фосфата и рибозы и протонами амино групп. Следовательно, в реальных условиях постоянно присутствующих протонов около атомов кислорода и азота нет. Нет также и постоянно присутствующих в указанных местах гидратации молекул воды. Однако наличие обмена протонами и молекулами воды между гидратированным нуклеотидом и растворителем (водой) следует трактовать как факт, свидетельствующий о вероятности пребывания протонов или молекул воды в соответствующих местах нуклеотида. Это будет служить препятствием для подхода к указанным местам других молекул, например, самих же нуклеотидов, т.е. мы встречаемся в данном случае с понятием динамического стерического препятствия. Это понятие, по нашему мнению, лучше отражает реальное существование молекул АМФ в воде. Следует учитывать также данные о вращении отдельных частей молекулы: фосфатной группы вокруг экзоциклической связи С4- С5и гетероцикла вокруг гликозидной связиC-N. Предполагаемая синхронность вращения этих групп позволяет в дальнейшем при рассмотрении ассоциации использовать представления о сотоянии АМФ с более вероятным транс расположением фосфатной группы и анти ориентацией гетероцикла. Другими словами, межмолекулярные соударения молекул АМФ наиболее вероятны именно для указываемого состояния этих молекул. Решению сложной задачи определения геометрического расположения осно
46
ваний АМФ при ассоциации должно способствовать представлению АМФ в таком виде, когда динамические препятствия как-то определены. Как нам кажется, такое представление о молекуле АМФ в воде (бесконечное разбавление, нормальные условия) следует рассматривать как наиболее правильное. С точки зрения анализа данного состояния АМФ проведём попытку объяснения наблюдаемой последовательности ассоциаций и явлений, сопутствующих образованию агрегатов. Наиболее доступной областью для подхода других молекул к АМФ в нашем рассмотрении оказывается пространство между Н-2 и Н-8.Не случайно, что при ассоциации нуклеотидов эти протоны всегда оказываются подверженными действию кольцевых токов соседних гетероциклов. Наиболее вероятной структурой димера должно быть такое взаимное расположение гетероциклов, которое соответствует минимальному нарушению динамических стерических препятствий и максимальному перекрытию стерически доступных областей. В данной структуре сильно гидратированные рибозо-фосфатные остовы максимально удалены. Шестичленный цикл одного нуклеотида находится ближе к пятичленному циклу другого основания, что приводит, согласно нашим данным, к специфическому для димеров соотношению изменения химических сдвигов протонов Н-2 и Н-8 [27].ГруппыNH2в гетероциклах направленывпротивоположные стороны, поскольку в аденине с ними должно взаимодействовать большее количество молекул воды, чем с другими возможными центрами образования водородной связи, т.е. динамические стерические препятствия для них выше. Указанная модель хорошо согласуется также с современными данными по применению внутримолекулярного эффекта Оверхаузера в растворах нуклеотидов[197].Последующее
47
увеличение стопки нуклеотидов не может сохранить состояние удалённости рибозофосфатных остовов,поскольку их вращение охватывает большое пространство и представляет собой сильное динамическое стерическое препятствие при наложении других нуклеотидов. Поэтому последующее образование ассоциатов должно происходить с нарушением структуры димера. Получение три- меров вполне возможно, однако, в этом случае при максимальной удалённости фосфатных групп, что осуществляется при лучеобразной структуре тримера с основаниями в центре, симметричное относительно взаимодействия между гетероциклами молекул АМФ построение тримера оказывается невозможным. Такой тример не должен быть стабильным, так как одно из трёх пар взаимодействий между основаниями в тримере не является выгодным вследствие несоответствия предлагаемому взаимодействию в димере. Симметричное построение можно осуществить в случае тетрамера, т.е. придти к структуре, предлагаемой нами на основании экспериментальных данных. Учитывая, что в тетрамере существует влияние кольцевых токов гетероциклов как на протоны Н-2,так и на протоныН-8. симметричное расположение отдельных нуклеотидов должно быть таким, чтобы протоны Н-2 и Н-8 находились относительно соседних оснований примерно в одинаковых условиях. Кроме того, наличие динамических стерических препятствий в ри- бозо-фосфатных звеньях должно приводить к максимальному удалению, а соседние основания должны взаимодействовать также как в димере.
Схема образования тетрамера из димеров несколько видоизменяется, а именно, при столкновении димеров каждый из них “растаскивается”вследствие удаления фосфатных групп, что при
48
водит к расположению оснований в центре. При этом рибозо- фосфатнамчасть молекул образует гидрофильную оболочку.
Таким образом,структура ассоциатов пространственно сводит все амино группы нуклеотидов воедино. Скорее всего,гидрат- ные оболочки амино групп также объединяются, и в ассоциате получаются три слоя. Внешний - гидратированные фосфатные группы и рибозныеостатки,средний - менее гидратированные основания нуклеотида и внутренний - объединённая гидратная оболочка всех амино групп. Внутренняя область по мере увеличения числа мономеров в ассоциате должна также увеличиваться и, вероятно, может включать в себя не только молекулы воды, но и более крупные гидрофильные молекулы, например, неорганический фосфат или фосфатные остатки нуклеотидов. Такая возможность в излагаемом представлении хорошо объясняет указанные нами ранее необычные факты взаимодействия фосфатных и амино групп в водных растворах [28]. Предложенная структура ассоциатов АМФ позволяет объяснить также появление в спектрах ЯМР линий поглощения протонов амино групп нуклеотидов в воде[184]. Ранее сделанное нами замечание о возможности влияния образования ассоциатов на гидрофобное окружение амино групп в рассматриваемом представлении может быть подробно истолковано. Внутренняя оболочка ассоциата, в которой содержатся амино группы, окружена сравнительно гидрофобной средней оболочкой из гетероциклов. Поэтому проникновение молекул воды из внешней оболочки во внутреннюю оказывается менее вероятным процессом. Следовательно, атака молекул воды на амино группы во внутренней оболочке ассоциата должна быть значительно ослаблена.В этих условиях взаимодействие амино групп между собой,(межнуклеотидные водородные
49
связи )или взаимодействие амино групп с попавшим во внутреннюю оболочку фосфатом может вполне конкурировать с процессом гидратации амино группы. Поскольку переход от гидратированного состояния амино группы к комплексу с другой амино группой или фосфатом должен осуществляться через негидратированное состояние амино групп, концентрация которых мала, то а соответствии с указанным в [28] механизмом время жизни протонов амино группы в комплексе должно быть велико, т.е. линияNH2- группы может быть наблюдаема в спектре ЯМР водных растворов АМФ. Согласно принятому представлению наблюдение возможно при наличии ассоциатов,т.е. при достаточно высокой концентрации нуклеотидов, что подтверждается на опыте [184]. Обратим ещё раз внимание на сущность динамического стерического препятствия. В отличие от обычных стерических препятствий, которые мешают подходу любых молекул, динамические препятствия отталкивают не всякие молекулы. В нашем случае в водных растворах обмен молекулами воды между растворителем и гидратной оболочкой отдельного фрагмента молекулы препятствует подходу гидрофобных остатков или молекул с меньшей способностью образовывать водородные связи, чем данный фрагмент. Ориентация молекул в ассо-циате должна стремиться к такому состоянию, когда во внешней оболочке будут располагаться группы, к которым больше всего“стремится” растворитель, т.е. группы, которые легче всего образуют водородные связи с растворителем (максимальный энергетический выигрыш при взаимодействии с растворителем). Ориентация остальных групп во многом определется этим фактором, хотя их внутреннее взаимное расположение также зависит от степени связывания с окружающей средой. При наличии разных по степени
50
взаимодействия с растворителем оболочек в ассоциате указанную ориентация можно объяснить при помощи физических терминов. Если обозначить энергетический выигрыш при взаимодействии отдельной группы с растворителем (Е), то очевидно, что в ассоциате существует градиент Е и соответственно ориентирующая силаF=dE/dr,гдег-направление максимального градиента энергии. Поскольку ориентационные силы возникают в таком представлении только при образовании ассоциата, то остаётся ещё невыясненным вопрос о сущности тех сил, которые ьы называем гидрофобными и которые обуславливают притяжение отдельных нуклеотидов. Согласно данным гл.2 донорно-акцепторные взаимодействия между нуклеотидами не являются определяющими при образовании ассоциатов. Причиной их образования оказывается взаимодействие нуклеотида с растворителем. Проблема определения геометрии подхода друг к другу ароматических радикалов нуклеотидов в воде является сложной вследствие наличия многих факторов, влияющих на“стэкинг”- взаимодействие,т.е. с точки зрения развиваемых представлений вследствие факторов, влияющих на динамические стерические препятствия в нуклеотидах. Перспективным способом получения данных о геометрии вторичных структур оказалось изучение внутримолекулярных взаимодействий в производных нуклеотидов, содержащих остаток ароматической аминокислоты.
2-6. Исследование взаимного расположения ароматических циклов в нуклеотид ил-(5-N) - аминах производили с учётом предполагаемого ранее [29,137] и подтверждённого в наших совместных работах [76,78] гидрофобного характера их взаимодействия. Факт преимущественной анти ориентации гетероциклов в нуклеотидамидах [78,79] также был использован для выработки
51
представления о наиболее вероятном состоянии этих молекул в растворе. Для построения конформационных моделей нуклеотидов но данным ПМР спектроскопии были использованы следующие предпосылки.
1.Протонизация основания должна вызвать сдвиг сигналов близлежащих групп в сторону слабого поля. Степень влияния этой своеобразной“зарядовой метки” на протоны зависит от расстояния
и, следовательно, даёт информацию о внутримолекулярной структуре,
2.Увеличение температуры, ускоряя вращение внутримолекулярных групп, изменяет также величины химических сдвигов всех внутримолекулярно взаимодействующих групп, т.е. указывает на взаимосвязь, формирующую структуру. Основным условием использования этой предпосылки служит выбор достаточно малой концентрации нуклеотидамида,исключающей межмолекулярное взаимодействие [39,80].
3.Анализ величин разности химических сдвигов сигналов от протонов нуклеотидамида и сигналов от соответствующих протонов нуклеотида и аминокислоты позволяет построить геометрическую модель молекулы на основании оценки расстояний между группами. Справедливость последнего утверждения следует из известных теоретических работ Джонсона и Пюльмана [136,145]. Первые положения(в применении к нуклеотидамидам) требовали дополнительных физико-химических обоснований.
2-7. Влияние ионизации на внутримолекулярное комплек- сообразование в аденилил- (5/V-N)-n-анизидинсЛ1ри наличии в растворе смеси развёрнутой и свёрнутой форм константы иониза
52
ции каждой из них могут не совпадать вследствие разной способности ионизированной и не ионизированной форм образовывать внутримолекулярный комплекс.
В результате тщательного исследования нами показано [38],что при протонизации основания константа свёртывания аденилил-(5У-N)- анизидина увеличиваетсяК+=3,6 К+2,6. Полученные данные об увеличении степени свёртывания при ионизации свидетельствуют об усилении взаимодействия между основаниями,которое должно приводить к изменению вторичной структуруры молекул полинуклеотидов.
2-8. Определение термодинамическиххарактеристик внутримолекулярного комплексообразования. Нами на основании температурной зависимости химических сдвигов разработана методика определения термодинамических величин внутримолекулярного комплексообразования аденилил - (5/-N)- п - анизидина [40].В отличие от от межмолекулярных взаимодействий определение термодинамических характеристик внутримолекулярных процессов было разработано недостаточно. Анализ выше указанных температурных зависимостей привёл к необходимости допущения внутримолекулярных комплексов двух типов: комплекса, не изменённого под влиянием растворителя, и комплекса с участием растворителя. Величина изменения энтальпии между ними равна 3,4 ккал/моль,т.е. переход комплекса из формы с близко расположенными основаниями(большое влияние кольцевых токов)в скошенную форму происходит с образованием внтримолекулярной водороднойсвязи.
53
Физико-химический анализ различных внутримолекулярных процессов в аденилил-^7-N)- п - анизидине позволил подойти к построению геометрических моделей нуклеотидамидов.
2-9. Построение геометрических моделей нуклеотидамидов.
Следует подчеркнуть, что без физико-химических исследований конформационных изменений в аденилил- (5/-N)- п - анизидине создание общей картины взаимного расположения отдельныхI'pynnв данной молекуле представлялось бы не строгим.Цель детального изучения аденилил-(5 -N)- п - анизидина состояла в том, чтобы на этом примере убедиться в правильности применения выше изложенных предпосылок и в дальнейшем для других нуклеотидамидов пользоваться только некоторыми из них. Выбор данного соединения был сделан вследствие наличия максимального по сравнению с другими нуклеотидамидами эффекта сближения ароматических циклов, наблюдаемого в спектрахKD[29,137] и в спектрахЯМР[39]. Качественная картина взаимного усреднённого расположения групп в аденилил-^-N)-п-анизидине предполагает наклонное положение плоскости анизидина и плоскости аденина с последовательно удаляющимися от основания группой ОСНз, мета и орто протонами кольца анизидина. Получив качественное представление о внутримолекулярной структуре аденилил-(5/-N)- п - анизидина,оказалось возможным перейти к количественным характеристикам и получить полную геометрическую картину расположения отдельных групп в этой молекуле,что намиибыло сделано в работе [80].
54
Предложенный способ построения конформационной модели аденилил-^-N)- п-анизидина был применён при создании качественных геометрических моделей других нуклеотидамидов. Для получения качественной картины пространственного расположения ароматических циклов в аденилил-^-N)-аминокислотах нами были использованы данные температурных и pH зависимостей величин химических сдвигов линий поглощения протонов отдельных группмолекул, а также экспериментальные и расчётные данные о влиянии кольцевых токов ароматических систем на электронное экранирование протонов близлежащих групп [81]. Вопрос о систематизации качественных геометрических моделей относительного расположения ароматических циклов нуклеотидил-^-N)- аминокислот был решён в нашихсовместных работах на основании только экспериментальных и расчётных данных о влиянии кольцевых токов ароматических систем на электронное экранирование протонов близлежащих групп [76]. Получено четыре типа взаимного расположения отдельных групп в нуклеотидил-(5/-N)-аминокислотах. К1-му типу относятся фенилаланиновые и тирози- новые производные пуриновых нуклеотидов; ко 2-му типу - трип-тофановые производные пуриновых нуклеотидов; к3-му типу - фенилаланиновые и тирозиновые производные пиримидиновых нуклеотидов и к 4-му типу - триптофановые производные пиримидиновых нуклеотидов. Одним из результатов наших исследований
[76,81] явилось обнаружение различия внутримолекулярных структурDиLформ нуклеотидил-(5;-N)-аминокислот(сd- рибозой), которое проявляется в спектрах ПМР для всех соединений. Спектры ПМРDиL- аминокислот илиN- (метилфосфо) -D-(L) - аминокислот не различаются. Механизм процессов, приводящих к
55
разным спектрам ПМР диастереомеров(в данном случаеDиL- нуклеотидил-(5/-N)-аминокислот) в ахиральных растворителях хорошо известен [56,70]. На основании данных ПМР нами предположено [81],что вD- формах ОСН3- группа расположена ближе к центру ароматического цикла аминокислоты чем в случаеL-форм,что соответственно приводит к другому расположению ароматического остатка аминокислоты и нуклеотида.
Построение геометрических моделей нуклеотидил-^-N)- аминокислот и их сравнение с моделью аденилил-(5/-N)- п - анизидина приводит к выводу о резком различии данных структур. Если в адснилил-(5‘-N)-п-анизидине ароматические кольца расположены практически взаимно перпендикулярно, то в адениловых производных нуклеотидамидов расположение ароматических систем ближек параллельному. Поэтому встал вопрос об исследовании зависимости нековалентных взаимодействий ароматических циклов нуклеотидамидов от расстояния между ними, т.е. от единственного существенного фактора, различающего эти соединения.
2-10. Зависимость нековалентных взаимодействийароматических циклов нуклеотидамидов от расстояниямежду ними.
Были исследованы аденилил-(5/-N)-w-аралкиламины с разным количеством метиленовых группп=1,2,4,6между ароматическими циклами [78].
Последовательный анализ изученных соединений по мере увеличения количества метиленовых групп позволяет понять закономерности, которые проявляются при сближении ароматических систем. Нами высказано предположение о том, что в этих нуклео-тидамидах ароматические циклы могут быть расположены в парал
56
лельных плоскостях, отстоящих друг от друга на 3,44 А°.По мере увеличения метиленовых групп фенильное ядро как бы “скользит”по плоскости аденина, удаляясь от протона Н-8 и приближаясь кNН2- группе. При этом расстояние от фенильного остатка до Н-2 протона почти не меняется. Небольшое увеличение действия кольцевых токов аденина на экранирование Н-2 может свидетельствовать об оптимальном расположении ароматических систем в аденилил~(5‘'-N)-2” -фенилэтиламине, который по расстоянию от фенильного радикала до фосфоамидного узла служит аналогом аденилил-(5-N)-фенилаланина, т.е. в нуклеотидил-^-N)- аминокислотах подход ароматических аминокислот к адениновому основанию может быть близок к межмолекулярному подходу отдельных компонентов.
Выше приведённые выводы соместных исследований
[76,78,81] позволяют сделать заключение о способах подхода ароматических молеул к гетероциклам нуклеотидов и, прежде всего,к адениновому основанию. Согласно полученным данным ароматические циклы в основном стремятся к области С$ - С6атомов углерода аденина. Можно предполагать также подход ароматических систем к области вблизи линии, соединяющей атомы Н-2 и Н-8 аденина, поскольку это также приведёт к указанным моделям.
Выяснению роли гидрофобных областей аденинового основания при нековалентном взаимодействии с ароматическими соединениями способствовало исследование внутримолеулярных процессов в молекуле дезоксиаденилил-(5/-N)- п - анизидина.
57
2-11.Сравнение внутримолекулярныхпроцессов, происходящихв рибо- и дезокснрибопронзводных молекулы аденилил-(5^Л)~II- анизидина[41]. Рассмотрение термодинамических характеристик внутримолекулярного комплексообразования в указанных молекулах позволяет предложить механизм свёртывания. Отрицательная величина изменения энтальпии внутримолеку-
г
лярного комплексообразования дезоксиаденилил-(5'-N)- п - анизидина(- 3,4 ккал/моль) свидетельствуют о преимущественном участии донорно-акцепторных взаимодействий в стабилизации данного комплекса. Согласно геометрической модели аденилил-^-N)-п- анизидина расположение плоскостей аденина и анизидина в комплексе близко к взаимно перпендикулярному, причём кольцо анизидина расположено над атомом кислорода рибозного цикла. Поэтому следует предположить, что полученные термодинамические величины относятся к взаимодействию атома кислорода рибозы с пи - электронами анизидина. В этом случае становится понятным факт необычного расположения ароматических систем в аде- нилил-(5/-N)- п - анизидине,которое обусловлено, с одной стороны,непосредственным соединением анизидинового кольца с фос-фоамидной связью, что ограничивает свободу перемещения в комплексе, а, с другой стороны, наличием указанных донорно-акцепторных взаимодействий.При гидрофобном взаимодействии ароматические циклы стремятся к максимальному перекрыванию, т.е. в большинстве случаев располагаются почти параллельно [192].
2-12. О природе различия дезокси и рибо производных аденозинфосфатов. Существенным различием молекул аденилил- (5'-N)- п - анизидина и дезоксиаденилил-(5/-N)- п - анизидина согласно данным по изучению температурных зависимостей вели
58
чин химических сдвигов является дополнительный процесс внут- римолекулярногопреобразования в аденилил-(5;-N)- п-анизиди- не, которое, как указывалось выше, вероятно, связано с образованием водородной связи. Поскольку к этому различию приводит появление ОН - группы в2/- положении рибозы, то очевидно следует предполагать образование водородной связиN3...Н-ОС2,стабилизирующей адениновыйцикл в несколько отличном от анти расположении. Подобное образование водородной связи могло быть и в молекуле АМФ. Однако температурные зависимости величин химических сдвигов протонов Н-2 и Н-8, полученные при исследовании перехода син-анти, не показывают наличия этого процесса. Данные факты свидетельствуют о том,что в отличие от свободного подхода молекул воды к атомамN3и протону группы НО-С2в АМФ появление гидрофобного ароматического цикла анизидина вблизи этих центров образования водородной связи с растворителемзатрудняет подход к ним молекул воды и способствует образованию внутримолекулярной водороднойСВЯЗИN3...НО-С2■
Обнаружение подобного факта образования дополнительных внутримолекулярных водородных связей позволяет сформулировать возможную роль С2- ОН группы рибозы при образовании вторичных структур, стабилизированных стэкинг-взаимодействием. Вероятно, изменение гидрофобности вблизи оснований нуклеотидов приводит к изменению процессов гидратации различных центров молекулы и даёт возможность осуществляться внутримолекулярным образованиям водородной связи, которые не могли происходить из-за конкурирующего взаимодействия активных мест связывания с молекулами растворителя.
59
Обобщая приведённые выше результаты, можно утверждать, что изучение внутримолекулярных взаимодействий в нуклеотида- мидах ставило своей целью получить данные о расположении ароматическихрадикалов вблизи аденинового и других оснований.
Получены пространственные модели аденилил-(5/-N)- аминокислот, где в качестве остатка аминокислоты выбраны фенилаланин, тирозин и триптофан.Главным моментом в представленных картинах взаимного расположения групп является взаимное расположение циклов аденина и аминокислоты. Последующие упрощённые“плоскостные”модели взаимного расположения гетероциклов других нуклеотидов и ароматических аминокислотпостроены в соответствии с опытом изучения серии производных адениловой кислоты.Модели необходимы для сопоставления аде- нилил-(5'-N)-аминокислот с другими нуклеотидил-(5/-N)- аминокислотами. Такое сопоставление способствует выяснению принципов формирования вторичных структур, включающих аде- нозин-5/-фосфат. Этой же задаче посвящено исследование зависимости взаимодействия ароматических циклов аденина и бензола от числа соединительных метиленовых групп.
Таким образом, изучение внутримолекулярных взаимодействий в нуклеотидам идах дало обширную информацию о геометрии различного окружения аденозинфосфатов, которую можно использовать при изучении более сложных структур, например, аденилил- (5/-З1')- аденилил-(5''-N)- фенилаланина [204]. Насколько достоверно полученное в результате исследования вторичной структуры аденилил-^-N)-аминокислот представление о подходе ароматической аминокислоты к аденину, которые являются ковалентно
60
связанными, должен был решить анализ соответствующих межмо- лекулярных взаимодействий.
Рассмотрение взаимодействия ароматических радикалов аминокислот и гетероциклов нуклеиновых кислот в более усложнённом виде оказывается вполне обоснованным,если ставить задачу нахождения характеристик элементарного комплексообразования нуклеотида и аминокислоты. Согласно литературным данным такая задача была не только не решена, но за исключением [118] в работах даже не принималось во внимание многообразие возможных взаимодействий в подобных системах, хотя с точки зрения выяснения специфичности связывания с нуклеиновой кислотой белков, содержащих ароматические аминокислоты, необходимо знать термодинамические и другие характеристики элементарного комплексообразования аминокислота - основание нуклеотида. Кроме того, если говорить о выяснении специфики подхода ароматических аминокислот к отдельному нуклеотиду(например, к аденозин-5/- фосфату),то в этом случае также необходимы данные об элементарном комплексообразовании.
2-13. Межмолекуляриые взаимодействия нуклеотидов и ароматическихаминокислот. Рассмотренные результаты по изучению внутримолекулярных взаимодействий радикалов ароматических аминокислот и гетероциклов нуклеотидов помогли представить геометрию их взаимного расположения и возможные внутримолекулярные процессы. Однако, термодинамика внутримолекулярного комплексообразования осталась невыясненной. Для решения вопроса о способах подхода ароматических аминокислот к нуклеотиду необходимо получить термодинамические параметры их межмолекулярного взаимодействия. Это осуществлено нами в
61
системах аденозин-5;- фосфат - ароматические аминокислоты [42,43].
Получение констант равновесия комплексообразования оснований нуклеиновых кислот и ароматических аминокислот представляет собой трудную задачу, решение которойс применением известных способов расчёта в ряде случаев оказывается неоднозначным |L18 ].Эта трудность обусловлена двумя причинами. Во- первых, в подобных системах имеются три типа взаимодействий: основание - основание, основание-аминокислота и аминокислота - аминокислота. Поэтому общее выражение для измеряемого параметра(наблюдаемого химического сдвига) должно включать три вклада, т.е. оказывается довольно сложным. Во-вторых, исследование самоассоциации ароматических систем представляет также большую проблему, в которой решается только задача нахождения механизма первичных стадий ассоциации. Осуществление расчётов межмолекулярного взаимодействия гетероциклов и ароматических аминокислот с использованием схемы последовательной самоассоциации нуклеотидов с равными ступенчатыми константами равновесия[118 Jявляется неоправданным, поскольку механизм полиассоциации в общем случае оказывается гораздо сложнее [27].
Нами обсуждены и выбраны условия, которые необходимы для изучения элементарного взаимодействия между двумя различными ароматическими циклами и для подобных систем предложен метод определения констант равновесия комплексообразования. Вначале подробно было изучено взаимодействие аденозин-5/- фосфата (АМФ) с метиловым эфиром тирозина (ТирОМе) в воде. При добавлении тирозина к АМФ происходит распад димеров АМФ и образование комплексов ТирОМе - АМФ. Когда влияние
62
распада димеров АМФ преобладает над образованием комплексов, химический сдвиг меняется в сторону слабого поля, затем по мере увеличения концентрации тирозина влияние образующихся комплексов становится основным и химический сдвиг меняется в сторону сильного поля. Подобное явление конкуренции димеризации и комплексообразования уже встречалось в наших работах ранее и было количественно детально нами описано [44].Наличие максимума характеризует одинаковое влияние двух разных процессов. Интересно, что в этой точке равновесная концентрация АМФ не зависит от константы комплексообразования, а полностью определяется контантой димеризации АМФ. Используя данные по влиянию на протоны тирозина кольцевых токов аденина [150],можно полагать, что центр ароматического кольца тирозина находится над центром аденина, причём группа СН2направлена в сторону шестичленного цикла аденина.
По аналогии с данной системой было исследовано межмоле- кулярное взаимодействие в воде аденозин-5/- фосфата с фенилаланином и триптофаном. Как видно из приведённых выше данных исследование внутримолекулярного взаимодействия ароматических аминокислот и нуклеиновых оснований в нуклеотидил-(5/-N)- аминокислотах позволило получить данные о взаимном расположении ароматических остатков в этих молекулах. Однако специфика подхода ароматических аминокислот к нуклеотиду, как уже отмечалось выше, может быть выяснена только после нахождения термодинамических характеристик комплексообразования ароматических аминокислот и нуклеотидов. Температурные зависимости величин химических сдвигов соответствующих внутримолекулярных комплексов в принципе могут дать подобную информацию. В
63
действительности же оказывается, что под влиянием температуры происходит не только раскрытие внутримолекулярного комплекса, но также изменение других внутримолекулярных процессов [40]. Поэтому определение термодинамических характеристик образования внутримолекулярных комплексов нуклеотидил-(5/-N)- аминокислот оказывается затруднительным. Неизвестно также влияние ковалентной связи между основаниями и аминокислотами на термодинамические характеристики их взаимодействия.
Разработанный нами метод определения констант равновесия межмолекулярного взаимодействия ароматических молекул [42] позволил получить термодинамические характеристики элементарных процессов комплексообразования нуклеотидов и ароматических аминокислот и определить последовательность вероятностей подхода ароматических аминокислот к нуклеотиду. Поэтому аналогично изучению ситемы АМФ - метиловый эфир тирозина были исследованы системы АМФ - фенилаланин и АМФ - триптофан,(см. табл. 1). Таблица1.
Константы равновесия комплексообразования АМФ с ароматическими аминокислотами.
СистемаКонстанта равновесия
АМФ - фенилаланин 27 л/моль
АМФ - метиловый эфир тирозина 45 л/моль
АМФ - триптофан 67 л/моль
64
Можно полагать в соответствии с данными таблицы 1,что благодаря различию в константах равновесия происходит“распознавание” на элементарном уровне молекулой аденозин-5'-фосфата определённой ароматической аминокислоты. При увеличении количества ароматических циклов в каждой из взаимодействующих сторон общая константа равновесия окажется близкой по величине к произведению элементарных констант равновесия, и вероятность подхода на уровне более сложных структур перейдёт в выбор определённых соединений.
На рис. 4 представлена зависимость логарифма константы равновесия от величины обратной температуры для системы АМФ
- фенилаланин. Данная зависимость нелинейна, а величины изменений энтальпии (0,2 - 1,5 ккал/моль) и энтропии (14-50),определённые на отдельных участках кривой, оказываются положительными. Зависимость термодинамических характеристик взаимодействия АМФ с фенилаланином от температуры свидетельствует о том, что механизм образования комплекса оказывается специфичным для каждого значения температуры, т.е. зависит от состояния растворителя. Это свидетельствует о гидрофобном характере взаимодействия в изученных системах. Поскольку гидрофобное взаимодействие при увеличении площади сталкивающихся частиц должно усиливаться, то этим можно объяснить наибольшую величину константы равновесия для системы АМФ - триптофан.
65
1.9
О-
17

Рис.4 Изменение логарифма константы равновесия образования комплекса аденозин-5/-фосфата с фенилаланином в зависимости от обратной температуры.
66
Интересна причина различия во взаимодействии с АМФ одинаковых по площади ароматических циклов фенилаланина и тирозина.Согласно экспериментальным данным [43] влияние кольцевых токов аденина на ароматические протоны этих аминокислот примерно одинаковое. Более того, во внутримолекулярном комплексе как аденилил-(5/-N)-фенилаланина, так и аденилил-(51-N)- тирозина расположение над аденином циклов фенилаланина или тирозина практически совпадает [81]. Необходимо отметить также, что для межмолекулярного комплекса АМФ - метиловый эфир тирозина изменение величин химического сдвина протоновСН2- группы и протонов в орто и мета положениях кольца тирозина находится в той же последовательности, что и во внутримолекулярном комплексе[81]. Следовательно, можно говорить об аналогии внутри и межмолекулярныхазаимодействий, определяющих пространственное расположение циклов в комплексе, и, соответственно, ожидать для близких по структуре комплексов АМФ - метиловый эфир тирозина и АМФ - фенилаланин одинаковых констант равновесия. Возможным объяснением значительного увеличения константы образования комплекса АМФ - метиловый эфир тирозина может служитьстимулированное сближением ароматических систем образование внутрикомплексной водородной связи, включающей ОН-группу тирозина. Возможность такого стимулирования в аналогичных системах показана нами ранее [41]. Согласно структуре комплекса АМФ - метиловый эфир тирозина вероятно происходит образование водородной связи ОН - группы с одним из атомов азота аденина.
67
Таким образом в результате этих исследований следует говорить о гидрофобной природе взаимодействия и установлении последовательности вероятностей подхода к нуклеотиду в системах ароматическая аминокислота - аденозин-5/-фосфат.
2-14. Межмолекулярные взаимодействия аденозинфосфатов с пептидами, не содержащими ароматические аминокислоты. Взаимодействие нуклеотидов с неароматическими аминокислотами не приводит к значительным изменениям в спектрах ЯМР. Усиление эффекта связывания в этих системах наблюдается при увеличении количества мономерных единиц в одном из компонентов.
Из двух основных видов взаимодействия - гидрофобного “стэкинг”-взаимодействия и кулоновского (взаимодействие заряженных групп) при образовании комплекса нуклеотид - пептид “стэкинг”-взаимодействие оказывается основным при наличии большего количества ароматических аминокислот в пептиде, а при отсутствии или их незначительном количестве основным взаимодействием,стабилизирующим комплекс, является взаимодействие разноимённых заряженных групп. Известно, что зарядовые взаимодействия в ассоциатах белок - нуклеотид или пептид - нуклеиновая кислота проявляются в спектрах ЯМР только при наличии большого количества зарядов хотя бы на одном из компонентов, т.е. в данном случае, когда один из компонентов полимер. Это нетрудно понять, если учесть, что изменения в спектрах ЯМР отдельных компонентов будут происходить в том случае, если концентрация образующихся комплексов достаточно велика. Вероятно, элементарное взаимодействие пары разноимённо заряженных групп недостаточно сильное, чтобы привести к заметной концентрации
ассоциатов. Кроме того, взаимное влияние зарядов в комплексе не приводит к заметному изменению величин химических сдвигов отдельных групп в результате достаточно удалённого расположения заряда подошедшей молекулы по отношению к тем группам первой молекулы, протоны которых не обмениваются со средой. В отличие от взаимодействия разноимённо заряженных групп молекул, образующих комплекс, в процессе которого сами реагирующие группы обмениваются, как правило, протонами со средой и их непосредственное влияние на величины химических сдвигов своих протонов в спектрах ЯМР в обычных условиях не наблюдается,“стэкинг”ароматических циклов даёт возможность осуществить влияние кольцевых токов этих циклов непосредственно на ароматические протоны взаимодействующих компонентов. Поэтому необходимым условием обнаружения взаимного влияния молекул при изучении методом ЯМР связывания нуклеотида и пептида за счёт взаимодействия их заряженных групп является осуществление сближения в комплексе заряженных групп с необменивающимися со средой протонами. Очевидно,что в этом случае не должно быть такого расположения других заряженных групп одного из компонентов комплекса, которое приводило бы к противоположному влиянию зарядов на необ- мепивающиеся протоны второго компонента, т.е. влиянию, снимающему эффект основного заряда, участвующего в образовании ионной связи. Это естественное условие наблюдения кулонговского взаимодействия нуклеотидов и пептидов требует перед постановкой такого исследования представления о конформационном состоянии или данных о взаимном расположении отдельных групп как нуклеотида так и пептида.
69
Нам представлялось важным для выяснения роликулонов- ских взаимодействий в многообразии межмолекулярных процессов аденознфосфатов обнаружить и изучить подобное взаимодействие между пептидом и АМФ.
В качестве пептида нами была выбрана молекула глутатиона (трипептид), в которой при нормальных значениях pH присутствуют как положительно, так и отрицательно заряженные группы. Выбор глутатиона достаточно произволен за исключением того факта, что эта молекула непосредственно функционирует в биологических системах [84,86]. В отличие от конформации АМФ в растворе, данные о которой были нами получены ранее и рассмотрены выше,конформацию глутатиона в воде необходимо было подробно исследовать, чтобы определить свободные положительно заряженные амино-группы, которые наиболее подвержены взаимодействию с отрицательно заряженными фосфатными группами АМФ.
Доказательство существования молекулы глутатиона в свёрнутой форме, стабилизированной кулоновским взаимодействием концевых разноимённо заряженных амино и карбоксильной групп, осуществлено нами в работе [45].
Оценка значения константы равновесия комплексобразования глутатиона с АМФ с учётом всех существующих в данной системе процессов ассоциации была осуществлена,исходя из следующих предположений. Если уменьшение действия эффективного отрицательного заряда фосфатной группы АМФ происходит вследствие созданияэффективной положительной оболочки вокруг фосфата при комплексообразовании АМФ с глутатионом,то можно считать действие положительно заряженной сферы (оболочка вокруг фос
70
фатной группы должна быть близка к сферической) равным действию точечного положительного заряда, расположенного в центре сферы. В этом случае эффект комплексообразования фосфатной группы с глутатионом должен быть схожим с эффектом уменьшения заряда фосфата. Подход однозаряженной группы глутатиона соответственно приведёт к уменьшению примерно на единицу заряда фосфта. Следовательно, изменение величины экранирования протона Н-8 АМФ при комплексообразовании с глутатионом должно быть близко к изменению экранирования протона Н-8 при титровании фосфатной группы, значение которого было нами измерено(18 гц).Использование данного значения позволило оценить константу равновесия комплексообразования АМФ с глутатионом(0,23 л/моль).
Такая величина константы значительно отличается, например, от константы равновесия комплексообразования АМФ с ароматическими аминокислотами (45л/моль), т. е. взаимодействие между заряженными группами слабее “стэкинг”- взаимодействия. В качестве положительно заряженной группы в молекуле глутатиона может бытьNH3TилиNH2+- группа. В сответствии с полученным представлением о конформационном состоянии глутатиона в воде, на наш взгляд, более предпочтительной для взаимодействиясфосфатом оказываетсяNH2+- группа, поскольку в свёрнутой форме вблизи этой группы находится только одна отрицательно заряженная группа(NH3+- группа с двух сторон экранирована отрицательно заряженными карбоксильными группами). Если не исключать возможности взаимодействия сЫНз+- группой, то скорее всего такое взаимодействие должно происходить в развёрнутой форме молекулы глутатиона. Это утверждение эквивалентно тому, что взаи
71
модействие фосфата с NH3+- группой приведёт к разворачиванию молекулы глутатиона. Однако необходимость дополнительной затраты энергии на раскрытие внутримолекулярного цикла при взаи-м о дейтев и исNH3+- группой свидетельствует о большей вероятности взаимодействия фосфата сNH2+- группой.
Таким образом, результаты данного исследования указывают на реальность процессов связывания отдельных нуклеотидов и неароматических аминокислот и пептидов за счет кулоновского взаимодействия разноимённо заряженных групп. Вследствие того,что величины констант равновесия комплексообразования АМФ с неароматическими аминокислотами значительно меньше чем величины соответствующих констант в системах АМФ - ароматическая аминокислота, то, вероятно, следует предположить и различные биохимические функции этих взаимодействий. По нашему мнению, структура образующихся агрегатов, в первую очередь, должна определяться взаимодействием между имеющимися ароматическими циклами, направляющими упорядочивание частиц. После осуществления этого первичного расположения частиц в комплексах для точной взаимной ориентации молекул будут играть роль болееслабые взаимодействия, такие каккулоновские.
Исключением в предлагаемой последовательности взаимодействий является случай, когда число взаимодействующих разноимённо заряженных групп значительно больше числа контактов ароматических систем. Тогда определяющим фактором будеткуло-новское взаимодействие, поскольку константа суммарного связывания всех заряженных групп окажется больше константы равновесия суммарного образования контактов ароматических циклов.
72
Исследование взаимодействия положительно заряженных lpynnпептидов с отрицательно заряженной фосфатной группой АМФ дополняет общую картину нековалентных взаимодействий с аденозинфосфатами.
Предшествующие данные позволяют создать общее представление о состоянии аденозинфосфатов в воде и об их способности к нековалентным взаимодействиям с различными соединениями.
Выделим основные моменты в экспериментальных результатах, позволяющие описать реальную динамическую структуру аде-нозин^-фосфата, которая проявляется при подходе молеул, содержащих ароматические системы. Согласно геометрической модели аденилил-^-N)- п - анизидина плоскость анизидина перпендикулярна плоскости аденина и соприкасается с ней вблизи атома С - 6 гетероцикла.
В аденилил-(5/-N)-бензиламиде удлинение цепи, связывающей гетероцикл и бензол за счёт добавления одной группы СН2между атомом азота и ароматическим кольцом приводит к выходу ароматического кольца на гетероцикл над атомом С-6. Однако, эта промежуточная конформация ещё не позволяет накладываться ароматическимсистемам. Кольцо бензола находится как бы на поверхности эллипсоида (вернее, какого-то аналога эллипсоида вокруг основания нуклеотида, поскольку его поверхность совпадает с изоповерхностью кольцевых токов аденина, т.е. с такой поверхностью, на которой влияние кольцевых токов аденина одинаковое,.Последующее увеличение числа метиленовых групп способствует
73
выдвижению бензольного кольца на плоскость гетероцикла и дальнейшему его продвижению в направленииNH2- группы.
Геометрия аденилил-(5'-N)-аминокислот с ароматическими радикалами, с одной стороны, подтверждает результаты исследования конформации аденилил-(5;-N)-бензиламидов, а с другой стороны даёт дополнительные сведения о свойствах аденозинфосфата. Аденилил-(5/-N)-фенилаланин имеет конформацию, сходную с аденшшл-(5/-N)-бензиламидом, причём метилированная карбоксильная группа располагается над фенильным кольцом, предоставляя возможность осушествить непосредственный контакт ароматических систем. Подход фенилаланина при межмолекулярном взаимодействии с АМФ происходит в соответствии с представялениями о внутримолекулярных взаимодействиях в аденилил-(5;-N)-фенилаланине. Существенным добавлением в конформацию метилового эфира аденилил-(5/-N)-тирозина,которая фактически оказаласьидентичной с конформацией аденилил-(5/-N)-фенилаланина, служит образование водородной связи ОН - тирозина с атомом азотаN- 7 аденина, показанное в системе АМФ+метиловый эфир тирозина. Подход триптофана к аденину как внутри- молеулярно, так и межмолекулярно происходит вблизи атома С-6, причём триптофан стремится к аденину шестичленным кольцом.Пятичленпый цикл триптофана располагается над областью от Н-8 до С-6 аденина. Расположение плоскостей ароматических систем в комплексе АМФ - ароматическая аминокислота близко к параллельному. Однако мы не только не может утверждать о строго параллельном расположении ароматических колец в комплексе, но на основании экспериментальных данных должны признать практически во всех случаях наличие отклонений. Создаётся впечатление,
74
что област ь С-5 - С-6 аденина является как бы широкой “щелью”для подхода гидрофобных групп (в данном случае ароматических систем). Взаимодействующий с АМФ ароматический цикл больше по размерам чем область С-5 - С-6,поэтому мы наблюдаем только заход в эту“щель” части кольца. С физико-химической точки зрения это можно понять, если представить, что к центрам образования водородной связи в молекуле АМФ (в гетероцикле - атомы азота) подходят молекулы воды, которые не дают всей плоскости подходящего ароматического кольца расположиться на плоскости аденина. Так как в одласти С-5 - С-6 (а также вблизи линии Н-2 - Н- 8) не следует ожидать образования водородных связей с растворителем,то взаимодействующий с АМФ ароматический цикл в этом месте может дойти до соприкосновения с гетероциклом. Очевидно, что указанные факторы оказывают существенное влияние на ориентацию плоскостей ароматических систем.
Наличие метилированной карбоксильной группы аминокислоты влияет на взаимное расположение ароматических колец в комплексе. С одной стороны, это дало возможность указать на механизм различия в конформациях DиLпроизводных аденилил- (5У-N)-аминокислот, с другой стороны, данный вывод свидетельствует о лабильности взаимного расположения ароматических систем. Нельзя утверждать, что найденное взаимное расположение циклов в комплексе останется таким же при изменении окружающей среды. Даже при появлении вблизи комплекса ароматических систем в воде молекул, которые практически оказываются инертными по отношению к стыкующимся циклам, стыковка будет нарушаться, если присутствие этих молекул изменит гидрофобностьокружающей комплекс среды. Непосредственное действие молекул
75
воды на компоненты комплекса изменится и, кроме того, изменится соотношение конкурирующих процессов межмолекулярного образования водородной связи с молекулами воды и внутримолеулярно-го образования водородной связи. Данный факт, с одной стороны, объяснил природу различия рибо- и дезоксирибопроизводных аденозинфосфатов,с другой стороны, позволил раскрыть соотношение различных процессов при формировании вторичной структуры с участием аденозинфосфатов.
Согласно полученным в работе экспериментальным данным основным процессом,соединяющим в водном растворе молекулы аденозинфосфатов и другие ароматические системы в первичные агрегаты, является гидрофобное взаимодействие. Появление гидрофобных агрегатов молекул способствует протеканию внутри этих комплексов процессов образования водородной связи и, вероятно, донорно-акцепторных взаимодействий. Поэтому структура комплексов определяется как внешними, так и внутренними факторами.Внешние гидрофобные взаимодействия определённым образом направляют молекулы в агрегаты, внутренние процессы приводят к точному взаимному расположению молекул в комплексе. Для аденозинфосфатов направление гидрофобного взаимодействия молекул определено в основном областью С-5 - С-6 атомов аденина (а также, хотя и в меньшей мере,областью между Н-2 и Н-8 вдоль линии, соединяющией эти атомы). Упорядочивание внутри комплекса ароматических систем с АМФпроисходит за счёт образования водородных связейN3...НОС2,N7...HO-R(R- например, остаток ароматической аминокислоты), и, вероятно, за счёт донорно- акцепторных взаимодействий(N...Р или П...П взаимодействия). Указанная последовательность взаимодействий оказывается
76
полезной при рассмотрении самоассоциации АМФ. Появление неожиданной структуры тетрамера в виде“четырёхугольной звезды”, которая удовлетворяла экспериментальным данным, становится понятным с точки зрения стабилизации определённой структуры ассоциата внутрикомплексными взаимодействиями. В отличие от стопки из 4-х нуклеотидов (высота стопки примерно в два раза больше её диаметра), в которой дополнительной стабилизации структуры не происходит, в тетрамере типа“звезда”три образующих стопку гидрофобных контакта между основаниями заменяются на четыре более слабых гидрофобных взаимодействия между основаниями и, кроме того, возможна дополнительная стабилизация такого ассоциата за счёт образования четырёх внутрикомплексных водородных связей. В предложенной структуре тетрамера это можно осуществить за счёт водородных связейNH2-групп адениновых оснований. Наличие упорядочивающих внутрикомплексных воздействий,наряду с соотношением гидрофобных областей и центров образования водородных связей в молекулах может оказаться основным фактором, определяющим вторичную структуру в ароматических системах и,в частности, в полинуклеотидах,
Чередование направляющих и упорядочивающих взаимодействий позволяет понять принципы нековалентного образования биологических структур в воде, явления их самосборки и распада, саморегуляции определённого соотношения таких образований в растворе, и,наконец, принципы управления подобными нековалентными процессами.
Конечно, каждое из выше указанных положений потребует в каждом конкретном случае подробного исследования. Однако выделение основных моментов, позволяющих заранее представлять
77
принципиальный механизм изучаемых явлений, несомненно облегчит эту задачу.
ГЛАВА3
СТРУКТУРА И СВОЙСТВА АССОЦИАТОВ ВОДЫ
Прежде чем рассматривать экспериментальные данные, свидетельствующие о структурированном состоянии водной среды, необходимо указать, что отсутствие линейной зависимости логарифма константы равновесия от обратной температуры в системах АМФ+АМФ и АМФ+фенилаланин (рис.Зи рис.4) требовало создания таких представлений о водной среде, которые смогли бы объяснить столь необычные выше приведённые кривые. Поэтому до выяснения структуры воды нами было предпринято исследование принципиальных основ поведения молекул воды вблизи гидрофобных поверхностей, что, на наш взгляд, должно было пролить свет на явление гидрофобного взаимодействия. Макроскопическое представление о гидрофобном вытеснении растворителем, т.е. водой, частиц, не обладающих центрами образования водородных связей, в некие образования не давало конкретного механизма вытеснения и тем более не объясняло дальнодействия или притяжения на расстоянии гидрофобных частиц.
3-1. Возникновение ориентационныхполей в водных растворах.
Рассмотренная в предыдущей главе природа не ковалентных взаимодействий между основаниями нуклеотидов в водных раство-
78
paxимеет как до норно-акцепторный, так и гидрофобный характер. Однако сам процесс сближения ароматических циклов при некова-лентном комплексообразовании в водных растворах,(“стэкинг”- взаимодействие), происходит в основном за счёт растворителя, т.е. за счёт гидрофобных сил. Нами дано теоретическое обоснование возникновения подобных силовых полей и получено экспериментальное подтверждение расчётных формул.
Понятие гидрофобной поверхности подразумевает отсутствие на ней центров образования водородных связей (ЦОВС). Вследствие этого молекулы воды, подходящие к такой поверхности, будут отличаться от своего обычного состояния в глубине сетки водородных связей водной среды направленностью всех ЦОВС приповерхностного слоя в сторону от поверхности,чтобы реализовать образование водородных связей. Таким образом, возникает градиент концентрации ЦОВС по направлению от поверхности в глубину водной среды. Таких ориентированных ЦОВС в приповерхностном слое молекул воды окажется практически вдвое больше, чем в обычном состоянии в глубине водной среды, хотя общее количество ЦОВС, интегрированное в любой точке по всем направлениям,естественно остаётся одним и тем же.
Развитие подобных представлений приводит к необходимости рассмотрения основных понятий теории ориентационных полей. Пусть в ансамбль частиц, обладающих некоторым набором свойств для взаимного связывания, внедрится некая частица с площадью поверхности S,не обладающая указанными свойствами частиц ансамбля. В этом случае поверхностьSоказывается медио- фобной или в частном случае для жидкостей соответственно соль- вофобной. Отсутствие вблизи поверхности возможности для частиц
79
реализовать все взаимные столкновения и связи, характерные для равномерно распределённых в объеме частиц, приводит к проявлению всех свойств взаимного столкновения и связывания частиц лишь по одну сторону от поверхности, т. е. приводит как бы к повышению эффективной концентрациии всех видов частиц ансамбля и их образований примерно в 2 раза.
“Избыток”концентрации у поверхностиSвоздействует на окружающие частицы, приводя к соответствующей“поляризации облаков соударения”с этими частицами.
Очевидно, что в силу неизменности числа центров образования связей у каждой частицы “поляризация облаков соударений”для неё,вызывая с какой-либо стороны увеличение числа центров, приводит в остальной чатси к соответствующему уменьшению.
Таким образом,всегда при наличии такой поверхности должны возникать две области повышенной и пониженной эффективных концентраций, приводящих соответственно к двум типам ассоциативного взаимодействия с разными константами равновесия. Производя полный расчёт [46], нами были получены формула зависимости от расстояния изменения свободной энергии ориентации1моля частиц среды и выражение для силы взаимодействия между разными сферамиSiиS2на некотором расстоянииR.
Учитывая, что ориентационное поле ЦОВС в водных растворах вызвано гидрофобными поверхностями, целесообразно данный вид ориентационного поля называть гидрофобным полем. Соответственно полученные формулы для изменения свободной энергии и силы будут выражениями для гидрофобной силы и энергии гидрофобного взаимодействия. Целесообразность термина
80
“гидрофобность”следует из важной роли оснований нуклеотидов и ароматических остатков аминокислот в формировании гидрофобных свойств макромолекулярных образований в водных средах. Причём изменение степени гидрофобности за счёт площади поверхностей приводит к изменению степени ориентированности ЦОВС, передающейся от гидрофобной площадки в среду. Такого рода“гидрофобная волна” в своём распространении, несомненно, будет оказывать влияние на“стэкинг”- взаимодействие и другие виды гидрофобного взаимодействия, являясь определённым сигналом или переносчиком информации.
Для того, чтобы формулу для энергии гидрофобного взаимодействия можно было применить к расчёту“стэкинг”- взаимодействия, необходимо учесть, что в реальных взаимодействиях абсолютно гидрофобных компонентов практически не бывает.
В общем случае на поверхности взаимодействующих в водных растворах веществ присутствуют как центры образования связей с растворителем, так и сольфобные площадки. Нами для общего случая также выведены соответствующие формулы для изменения величинсвободной энергии в процессе ассоциации между веществом и растворителем [46].
Обработка данных по взаимодействию соединений с наличием как мест связывания, так и сольвофобных площадок согласно выведенным формулам позволила,наконец, полностью объяснить ход зависимостей, приведённых на рис.З и рис.4и получить величины разностей между изменениями энтальпии и энтропии образования ассоциатов растворителя и комплексов растворителя с веществом.Соответственно, для системы АМФ+АМФ это -39,7
кДж/моль и -117 Дж/моль К, а для системы АМФ+фенилаланин это -36,4 кДж/моль и -119 Дж/моль К.
Следует обратить внимание на значительные величины изменения энтальпии, составляющие примерно величину изменения энтальпии образования двух водородных связей. Этот факт,скорее всего, обусловлен природой ассоциации молекул воды, выяснение которой составило довольно обширный этап исследований.
3-2. Гидрофобная модель структуры ассоциатов молекул
воды.
Учитывая выше приведённые представления о гидрофобно- сти, нами был предложен кинетико-термодинамический подход к описанию процессов ассоциации молекул воды. Прежде всего подробный анализ полного процесса полиассоциации позволил нам вывести формулу ограничения количества молекул в ассоциате [47]. К х М(1=m(m+l)/2,где К - ступенчатая константа равновесия, М0- собственная молярная концентрация воды,m- количество молекул в ассоциате.
В этом соотношении вследствие конечных величин КиМ^должна быть также конечной и величинаш. При М0—55 моль/л для Н20 и К=10 следует ожидатьm=32. Однако, зная, что разветвление ассоциата воды по конечной молекуле может идти сразу по трём связям и,соответственно, вместо К надо вставить ЗК, получится, чтош=57. Следовательно, в общей схеме ассоциации молекул воды в результате равновероятного увеличения числа молекулв каждом образующемся ассоциате их максимально возможное количество окажется равным 57.
82
Таким образом, можно ожидать,что при такомmв линейно разветвлённых ассоциатах молекул воды в основном будет осуществляться образование водородных связей внутри ассоциата, т.е. происходить образование циклических структур.
На основании представлений о гидрофобности можно определить увеличение степени гидрофобности как уменьшение отношения числа свободных водородных связей какого-либо образования, в нашем случае ассоциатов воды,к общему количеству молекул воды в данном образовании. Поскольку ассоциация молекул воды в таком представлении соответствует увеличению гидрофобности, то для рассмотрения путей формирования молеулярных структур воды целесообразно ввести своего рода принцип максимальной гидрофобности.
Развивая указанный принцип как необходимость возникновения ассоциатов для уменьшения отношения числа свободных центров образования водородных связей (ЦОВС) кобщему числу молекул в ассоциате, в применении к циклам и далее к сферическим образованиям получим для мономера - 4; для димера -3;для линейного пентамера - 2,4;для циклического пентамера - 2; для шестициклического тетраэдрического каркаса из17молекул-1,4; для додекаэдра -1;для додекаэдрического тетраэдра из 57 -ми молекул (глобулы, состоящей из четырёх связанных в виде тетраэдра додекаэдров)-0,7.
В соответствиии с ограничением числа молекул в ассоциате, равным57, т.е. как раз для последнего ассоциативного образования, следует остановиться в усложнении структуры ассоциатов и проанализировать получившийся результат.
83
Любое последующее межглобулярное образование водородной связи будет постепенно уменьшать степень гидрофобности. Однако, согласно представлениям о гидрофобном взаимодействии как определяющем энергетически наиболее выгодное взаимное расположение молекул по максимуму перекрывания соприкасающихся площадей при отсутствии на них центров образования водородных связей, вхождение каждого из четырёх додекаэдров глобулы в гидрофобную впадину другой глобулы, с одной стороны, не даёт10-ти центрам образования водородной связи на поверхности додекаэдра образовать хотя бы одну связь, с другой стороны, определяет максимальное соприкосновение по площадям.
Необычность такой состыковки глобул воды позволяет расширить действие принципа максимальной гидрофобности, уточнив выше принятое отношение как отношение к общему числу молекул количества свободных ЦОВС, имеющих реальную возможность или вероятность образовать водородную связь в создаваемом окружении, а не просто свободных ЦОВС.Тогда в этом определении для системы взаимно входящих друг в друга глобул такое отношение вообще стремится к нулю. Данное состояние даже теоретически является законченным и,следовательно, реально наиболее устойчивым.
Общий вид предлагаемой структуры ассоциата молекул воды представлен на рис.5. Это тетраэдр, составленный из четырёх додекаэдров, каждый из которых имеет 12 пятиугольных граней, 30 рёбер,20 вершин (в каждой соединяются три ребра, вершинами являются атомы кислорода, ребром служит связь О-Н...О. Всего в тетраэдре додекаэдров 57 молекул воды,17из которых составляют тетраэдрический полностью гидрофобный (т.е. насыщенный че
84
тырьмя водородными связями) центральный каркас, а в четырёх додекаэдрах на поверхности каждого находятся по 10 центров образования водородной связи (ОНили О).
Подтверждение предлагаемой структуры ассоциатов молекул воды, согласующейся с наиболее принятой моделью Полинга, но являющейся более развитой и усовершенствованной тетраэдрической системой, формирующейся по совершенно новому неиспользуемому ранее критерию максимальной гидрофобности, было осуществлено в ряде дополнительных исследований. Одним из первых экспериментов, который должен был свидетельствовать о наличии ограниченных по числу молекул воды ассоциатов, был этап изучения температурных зависимостей показателя преломления водной среды. Несмотря на то,что эти данные получались ранее и давно уже вошли в соответствующие справочники, нам представлялось целесообразным более тщательно повторить эти опыты, поскольку трактовка и способы анализа экспериментальных кривых качественно отличались от существующих прежде.
3-3. Температурные зависимости показателя преломления водной среды. Обработка температурных зависимостей проводилась для процесса циклизации молекул воды, записанного в общем виде:
М =Мсв,гдеМсв-свёрнутая в циклы форма ассоциата, М
-разрушенное или отличное от упорядоченной свёрнутой формы состояние ассоциата.
Выбор столь простой схемы изменения количества водородных связей в воде от температуры обусловлен приведённым выше выводом о преимущественом образовании водородных связей
85

Рис.5. Модель ассоциата воды из 57- и молекул - тетраэдр из четырёх додекаэдров (“квант”). Каждый из додекаэдров имеет 12 пятиугольных граней,30 ребёр,20 вершин(в каждой соединяются три ребра, вершинамиявляются атомы кислорода, ребром служит водородная связьО-Н...О).Из 57-и молекул воды“кванта”17 составляют тетраэдрический полностью гидрофобный(т.е. насыщенный.четырьмя водородными связями)центральный каркас, а в четырёх додекаэдрах на поверхности каждого находятся по10центров образования водородной связи(О-Н или О).
86
внутри ассоциата, что и должно определять изменение показателя преломления.Согласно полученнымданным[47] значения для свёрнутой формыпсв= 1,338 и для мономераn=1, 286 существенно отличаются друг от друга и большая величинаПсВ.свидетельствует об упорядоченности свёрнутой формы. Рассчитанные же величины изменения энтальпии(21 кДж/моль) и энтропии(51 Дж/моль К) рассматриваемого процесса [47] близки к соответствующим параметрам образования и разрыва одной водородной связи. Следовательно, разрушение свёрнутой формы ограничивается разрывом всего одной водородной связи, т.е. правильнее говорить о нарушении основной свёрнутой в циклы формы, а не о её полном раскрытии или разрушении.
Хорошее совпадение расчётных и экспериментальных кривых свидетельствует о справедливости применяемой схемы и последующих рассуждений, однако не показывает, сколь большое количество молекул воды участвует в построении основной формы ассоциата.
С целью выяснения этого вопроса и для доказательства ут- вержэденияо построении ассоциатов воды по принципу наибольшей гидрофобности были предприняты исследования водных растворов солей, влияющих на ассоциаты или изменяющих основную форму ассоциата воды.
3-4. Концентрационные зависимости показателя преломления водных растворовKCI. Обработка экспериментальных зависимостей изменения показателя преломления от концентрацииKCIв воде[47] привела к следующим выводам:
87
1.Небольшое увеличение показателя преломления воды в присутствииKCIпо сравнению с исходным означает приближение структурного состояния воды к большему количеству свёрнутых форм,т.е. при добавленииKCIпроисходит стабилизация образующихся глобул.
2. Сравнительно большое количество молекул воды, находящихся под влиянием иона (800) никак не укладывается в представление о том, что молекулы воды специфическим образом упорядочиваются вокруг иона, соответственно уходя из определённых структур ассоциатов, что также не свидетельствует о нарушении глобул.Несуразность больших значений полностью убирается, если рассчитать количество глобул, ориентированных вокруг каждого ионаК+и СГ.Действительно, если разделить 800 на количество молекул в глобуле по предлагаемой модели (57), то соответственно получается значение порядка 14.Поделив на два (по числу ионов) получим 7,что находится в неплохом согласии с представлением о координации молекул растворителя с ионом в основном по осям координат, т.е. приближение к иону близкого к 6-ти количества координирующих частиц. Это рассуждение в какой-то степени подтверждает реальность наличия глобул. В этой работе у нас ещё не было данных о возможности образования ассоциатов из нескольких глобул по 57 молекул. Однако, одна из естественных трактовок наличия около иона такого количества молекул как 800 предполагает возможность существования неких громадных образований-клат-ратов по несколько сот молекул воды в каждом. В этом случае представляется интересным само значение 800,которое указывает на порядок количества молекул в предполагаемом стабильном образовании из молекул воды.
88
На обработанных разными способами экспериментальных кривых присутствуют детали, которые для уравнений, описывающих процессы ассоциации, оказываются просто случайными отклонениями. Однако, регулярность этих отклонений через определённый интервал (0,8 - 0,9%) и повторяемость во всех независимых опытах заставила провести эксперимент, надёжно улавливающий и фиксирующий указанные отклонения.
С этой целью в области концентраций KCI12-14% с шагом
0,1%с повышенной точностью как приготовления, так и измерения в разных условиях по температуре и давлению были получены зависимости показателя преломления от концентрацииKCI(рис.6). Как видно из рис.6, обнаружены действительно регулярные пикиуменьшения показателя преломления, причём разность между пиками по концентрацииKCIпо расчёту соответствует уменьшению среднего объёма между ионамиК+иCI" на объём одной молекулы воды. Так в изученном диапазоне (12-14%) число молекул в ассоциате с повышением концентрации уменьшается последовательно15-14-13. Иными словами, обнаружено замечательное явление“раздевания”глобулы по одной молекуле при увеличении ионной силы. Насыщенность раствора и соответственно полное разрушение глобул происходит при 22 - 24%KCI.
При понижении концентрации KCIинтервал концентраций на изменение объёма,равного объёму одной молекулы воды, быстро уменьшается, что,естественно, затрудняет его экспериментальное обнаружение.
Одним из выводов данного исследования служит также указание на изменение фазового состояния воды, поскольку ступеньки,
89

Рис.6. Зависимости показателя преломления (п) от
концентрации KCIв воде при21(1) и 25°С (2).
90
проявляемые в полученных зависмостях свидетельствуюто мякрофазовых переходах и, следовательно, о стабильности существующих в воде ассоциатов.
Подтверждение существования стабильных образований в воде удалось получить из данных протонного магнитного резонанса.
3-5. Исследование структуры воды методом протонногомагнитного резонанса [48]. В предлагаемой модели термодинамически устойчивого ассоциата -“додекаэдрического тетраэдра”из 228 центров образования водородных связей (ЦОВС) 188 связей, т.е. 94 протона,образуют практически неразрушаемый симметричный каркас ассоциата, оставляя на поверхности 40 центров возможного образования водородных связей, т.е. 20 протонов. Поскольку предполагаемая относительная неразрушаемость каркасаассоциата подразумевала отсутствие обмена молекул воды ассоциата с молекулами других ассоциатов, то в спектре ПМР в области поглощения ОН - группы теоретически предполагалось обнаружить две группы пиков в соотношении 94/20 с расположением более интенсивного пика в более слабом поле.
В опытах использована сверхчистая вода (18МОМ см), полученная на установке фирмы“Миллипор”. Спектры ПМР были получены на спектрометре “WM- 500Bruker”,Во избежание искажений в спектре мощность подаваемого импульса была выбрана относительно слабой, гарантирующей сохранение правильной формы сигнала. Вращение образца было исключено из-за возможного по
91
явления дополнительных боковых пиков. Необходимая для разрешения однородность поля достигалась за счёт выбора измеряемого образца в виде тонкого капилляра с водой, помещённого в фиксированное по центру стандартной ампулы положение, а также за счёт тщательной настройки однородности поляч соответствующими шимам и. На рис. 7 представлен спектр ПМР воды.В спектре присутствуют две группы пиков: основной синглет и группа близких по значениям химических сдвигов слабых пиков в диапазоне)0гц как в сторону сильного,так и в сторону слабого поля. Соответствием полученного спектра ПМР предложенной модели ассоциата из 57 молекул воды (рис.5) следует считать прежде всего отсутствиеюлько одного ожидаемого по обычным представлениям сигнала от протонов воды и появление большинства дополнительных пиков в более сильном поле.
Серьёзным подтверждением модели служит отношение площади основного пика к площади группы слабых сигналов. Согласно модели, это отношение должно быть равно 94/20,что очень близко соответствует отношению соответствующих площадей, взятых из спектра ПМР. Наличие дополнительных линий, тем более пика в более слабом поле, нетрудно объяснить при рассмотрении возможного реального поведения термодинамически устойчивого ассоциата (“кванта”) по взаимодействии с себе подобными структурами.
Действительно, из анализа модели додекаэдрического тетраэдра следует, что шесть ЦОВС на каждой грани тетраэдра пространственно по расположению и направлению связей точно соответствуют шести ЦОВС на грани любого другого тетраэдра. Естественно,тодля образования всех шести водородных связей между
92


Рис.7. Спектры ПМР(“Bruker-WM- 500”)протонов воды(1)и после16накоплений (2).
Расшифровка спектра ПМР на рис. 7.
На спектре ПМР стрелками показаны выявленные линии поглощения протонов воды. Анализ выделяемых из интегрального спектра 9-ти составляющих линий с соответствующими площадями позволил оценить долю протонов(f)в каждом положении по отношению к общей интегральной сумме площадей всего спектра. Ниже приведены полученные таким образом экспериментальные величины(f3KC)и для сравнения соответствующие теоретические величины(Гтеир.),рассчитанные из окончательной 912-ти молекулярной модели структурного элемента воды. Разделение протонов
93
по степени экранирования произведено в соответствии с моделью и согласовано с расположением линий в спектре ПМР.
Тип и количество протонов. f3KC
1.Свободные поверхностные протоны
на двух острых углах элемента (4).0,0022 0,0027
2. Протоны двух центров
супертетраэдров (8).0,0044 0,0051
3.Сферические и каркасные протоны
“квантов”(1152 + 288).0,7894 0,7836
4. Протоны на углах “квантов,”
направленных во внутрь (28 ).0,0154 0,0143
5.Свободные поверхностные протоны
на гранях элемента кроме центра (60).0,0329 0,0382
6. Протоны на связях, перпендикулярных
поверхности грани (72).0,0395 0,0440
7. Протоны в центре “квантов”кроме
центров супертетраэдров (56).0,0307 0,0294
8. Свободные поверхностные протоны
в центре грани (36).0,0197 0,0215
9. Протоны комплементарных связей
между “квантами”в элементе(120).0,06580,0612
94
гранями разных тетраэдров чередование -ОН и -О в центрах образования водородных связей на разных гранях должно быть взаимно дополняющим, т.е. комплементарным. Комбинаторный анализ чередования -ОН и -О в центрах образования водородных связей приводит к выводу о существовании четырёх типов комплементарных шестисвязных пар граней разных тетраэдров с соотношением интенсивностей 1:6:15:10. В спектре ПМР этот квартет линий по всей видимости наблюдается в виде уширенного сигналовв более сильном поле по отношению к основному пику. Этому предположению соответствует относительное количество протонов,отвечающих площади данного сигнала.
Расчёт по предложенной модели количества возможных шестисвязных комплементарных пар показывает значительно меньшую вероятность получения подобных пар уже на второй из четырёх граней тетраэдра. Поэтому одна комплементарная пара оказывается основной ячейкой в построении последующих ассоциатов (например,в результате образования водородных связей между пространственно соответствующими вершинами спаренных граней при подходе комплементарных пар друг к другу,т.е. по 4 и по 2связи).
Поворот комплементарной пары как наименее связанной ячейки в сетке водородных связей приводит вследствие расположения положительных и отрицательных зарядов водородной связи на различных радиусах вращения вокруг центра тяжести к появлению магнитного момента ячейкм, что позволяет объяснить наличие остальных линий в спектре ПМР.
95
Сигналы от протонов водородных связей, попадающих под экранирующее действие магнитного поля, окажутся в более сильном поле, а сигналы от протонов, наиболее удалённых от центра вращения, попадающих под дезэкранирующее влияние магнитного поля, окажутся в более слабом поле по отношению к основному сигналу. Количество, положение в спектре и относительная интенсивность всех наблюдаемых линий в принципе хорошо соответствуют предлагаемой модели, однако детельная расшифровка каждой из линий требует серьёзных джополнительных исследований, и потому полное объяснение линий на этом этапе не представлялось целесообразнывм.
Предложенная модель и предположения о соответствии с ней данных ПМР в этой стадии исследований носили, естественно, гипотетический характер и представляли интерес, скорее всего, для выбора последующих экспериментов,подтверждающих сугцест- воание в воде термодинамически стабильных структур. Тем не менее следует отметить,что наличие отдельных линий поглощения в спектре ПМР непосредственно и наиболее наглядно доказывает наличие стабильных ассоциатов в воде.
Обнаружение в спектрах протонного магнитного резонанса воды в области более сильного поля по отношению к основной линии поглощения фактически трёх областей химического сдвига с интенсивными линиями поглощения на общем нулевом фоне может объясняться либо наличием сложных структур одного типа ассоциатов,либо сущестованием разных типов образований из “квантов”воды.
96
Важная роль структурного состояния водной среды в жизнедеятельности организмов требовала серьёзного планирования дальнейших исследований и понимания того, что вопрос о возможной стабильности вполне определённых ассоциатов из молекул воды не поднимался, поскольку считалось, что малое время жизни комплексов с водородной связью (10'9сек.) исключает появление долгоживущих комплексов на основе этих связей.
Поэтому доказательство наличия своего рода фракций воды подразумевало обязательное подтверждение существования стабильных ассоциатов воды непосредственно в хроматографических исследованиях. Учитывая предшествующие данные о степени стабилизации термодинамически устойчивых “квантов” воды, нами был предпринят эксперимент по выяснению возможности разделения фракций воды методом жидкостной хроматографии [49].
3-6. Экспериментальное доказательство наличия фракций воды. Прежде всего следует отметить, что такие странные факты, как отклонение от линейной при снятии зависимости показателя преломления от процентного содержания в водном растворе ряда веществ: метиловогоспирта, ацетонитрила и других,- явно свидетельствовали о разрушении каких-то устойчивых образований из молекул воды.
Причём поражало также то, что это явление довольно специфично и, например, для метилсульфоксида линейность в аналогичной зависимости сохраняется, т.е. в этом случае со структурой воды как бы ничего не происходило. Это в какой-то мере подтверждается тем, что смесь диметилсульфоксид-вода, известная в медицине как“диметоксин”, используется для инъекций и, следовательно, не яв
97
ляется токсичной в отличие от водных растворов метилового спирта и ацетонитрила.
Выбор условий определения фракционного состава воды методом высокоэффективной жидкостной хроматографии.
Поскольку вода не поглощает в характерном для наиболеедоступных детекторов в диапазоне 200 - 700 нм и не обладает флуоресценцией, использование наиболее распостранённых и надёжных спектрофотометрического и флуорометрического детектора, на первый взгляд, представляется невозможным. Однако“прозрачность” воды в ультрафиолете позволяет прибегнуть к косвенному детектированию с использованием элюентов, поглощающих в УФ - диапазоне. К таким элюентам относятся ацетон, уксусноэтиловый эфир,бензол и т.п. Выбор предпочтительного растворителя определяется двумя факторами: выбором колонки и возможностью получения растворителя с минимальным содержанием воды. Детектирование при этом возможно по снижению поглощения (фона детектора) при прохождении пика воды. Возможно также использование рефрактометрического детектирования при достаточной чувствительности рефрактометра.
Как было уже показано нами [47], возможные фракции воды обладают различной гидрофобностью, следовательно для их разделения можно использовать обращённо-фазную хроматографию. Однакообращённо-фазное элюирование чистым органическим элюентом привело бы к элюции столь полярного соединения как вода практически в мёртвом объёме. В связи с этим для разделения было использовано обратное гидрофобности свойство фракций воды - различие в способности к образованию водородных связей.
98
Для разделения фракций была использована колонка с привитыми к силикагелю NH2- группами, обладающими заметной способностью к образованию водородных связей. Учитывая, что энергия образования водородных связей сопоставима с энергией дисперсионных взаимодействий,и что использование органических растворителей обеспечивает достаточно высокое удерживание на аминноЙколонке таких соединений, как моно- и дисахариды и спирты,NH2- колонка была выбрана для оценки возможностей разделения фракцийводы.
В качестве элюентов была оценена возможность использования при косвенном детектировании двух органических растворителей,поглощающих в УФ - диапазоне: ацетона и уксусноэтилового эфира(длины волн, соответственно,250 нм и 290 нм). В случае прямого рефрактометрического детектирования оценивали возможность использования этилацетата и ацетонитрила.
Результаты эксперимента при косвенном УФ детектировании. С использованием обоих элюентов удалось выявить элюциюводы сNH2- колонки тремя частично разделёнными хроматографическими зонами на фоне общего сильно размытого пика (рис. 8 а-б).
При этом использование ацетона позволяло получить более чёткие зоны, иначе говоря, эффективность колонки была выше. Постепенно происходило размывание хроматографических зон, что может быть связано с насыщением колонки водой (рис. 8в).
2
Рис. 8 а. Косвенноедетектирование фракций воды по изменению поглощенияэтилацетата на длине волны 290 нм.
8 б.Косвенное детектирование фракций воды по изменению поглощения ацетона на длине волны 250 нм.
8 в. Косвенноедетектирование фракций воды
по изменению поглощения этилацетата на длине волны 290 нм в стадии насыщения колонки водой.
100
Более тщательный анализ хроматографических зон на рис.8а, рис. 8бпоказывает, что правильнее трактовать наблюдаемые линии как две группы линий на общем размытом пике.
Так на рис. 8 а видно, что при использовании в качестве элю- ента этилацетата вначале отдельно выходит небольшой пик, а затем прослеживается группа линий, фактически представляющая собой неразрешённый триплет, ширина линий которого заметно больше, чем у первогопика.
Такое наблюдение и условное подразделение оправдывается анализом хроматографических зон на рис. 8 б.При использовании в качестве элюента ацетона,резко меняющего ширину линий, вначалеможно очертить отличающийся от остальных широкий пик, а
I
затем чётко видны три линии.Поэтому даже при предварительномкосвеном детектировании можно утверждать о наличии фракцийводы.
Использование прямого рефрактометрического детектирования. Использование косвенной УФ - спектрофотометрии не позволяло количественно проанализировать результаты разделения, поскольку оптические характеристики детектора в этом режиме отличались нестабильностью. Чувствительность метода была невелика, что вело к перегрузке колонки. Тем не менее полученные предварительные результаты, а также данные о показателях преломления воды с различными органическими растворителями позволили предположить, что использование рефрактометрического детектирования позволит улучшить условия разделения. В связи с этим на следующем этапе разделение воды на фракции методом выскоэф-
101
фективной жидкостной хроматографии проводили с детектированием по разности показателей преломления элюент - вода.
Поскольку при использовании косвенного УФ - детектирования хорошие результаты при разделении фракций воды былиполучены с использованием в качествеэлюента этилового эфира уксусной кислоты, в первоначальных экспериментах с рефрактометрическим детектированием был использован тот же элюент. Однако при этом не удалось обнаружить сколько-нибудь выраженных хроматографических зон или даже системных пиков.
В связи с этим на следующем этапе нами был использован широко применяемый в хроматографической практике апротонный растворитель -ацетонитрил. Практически полная прозрачность этого растворителя в УФ - диапазоне не позволила использовать его при разделении фракций воды с косвенным УФ - детектированием. В то же время ряд характеристик(низкая вязкость, полное смешение с водой)позволили предположить,что он может быть успешно применён в качестве элюента при разделении фракций воды с рефрактометрическим детектированием.
Полученные результаты полностью подтвердили это предположение. Методом ВЭЖХ на аминной колонке с использованием в качетсве элюента ацетонитрила удалось разделить на фрактции как чистую воду, так и её смеси с ацетонитрилом в различных соотношениях(рис. 9б- 9 е). При этом было обнаружено, что при различных соотношениях ацетонитрил - вода количество фракций и эффективность разделения различны. Например, при использовании чистой воды на хроматограмме наблюдаетсяпри первом рассмотрении три пика, соответствующие трём фракциям, а при ис
102
пользовании 50 - ти процентного раствора воды в ацетонитриле(объём на объём)- фактически только одна фракция(рис. 9 в).Дополнительная проверка показала, что при инжектировании чистого ацетонитрила наблюдается единственная хроматографическая зона, элюирующая в виде негативного пика с удерживанием около107 сек.(рис. 9 а - 1,78 мин.). Наличие этой зоны свидетельствует о том, что ранее отмеченные нами пики действительно нри-надлежат воде, и позволяет определить столь важный параметр как мёртвое время колонки, необходимое для дальнейших хроматографических расчётов.
Для понимания механизма разделения фракцийводы наNH2- колонке при использовании в качестве элюента 100 % ацетонитрила следует рассмотреть имеющиеся представления о сорбции на этой колонке представителей группы веществ, также способных к образованию нескольких водородных связей - углеводов. Поскольку аминные колонки и элюенты типа ацетонитрил - вода широко используются для хроматографического анализа различных представителей этого класса органических веществ, существуют как многочисленные экспериментальные данные о возможном механизме их сорбции, так и определённые теоретические представления, анализ которых может оказаться полезен при рассмотрении механизма хроматографического разделения фракций воды.
Известно, в частности, что с увеличением молекулярного весасахаров, их удерживание наNH2- колонке резко возрастает. Существует также определённая зависимость между удерживанием и содержанием воды в элюенте: чем больше процентное содержание воды, тем иеньше удерживание углеводов. Таким образом, механизм удерживания углеводов на аминной колонке существенно от
103

Рис. 9 а - Прямое рефрактометрическое детектирование. Базовая хроматограмма - время выхода мёртвого объёма в отсутствие порций воды.I-интенсивность, пропорциональная изменению показателя преломления.
б-Прямое рефрактометрическое детектирование фракций 100-процентнойводы.
в - Прямое рефрактометрическое детектирование фракций воды (50% в ацетонитриле).
104
).мл

__t
гз
Рис. 9 г -Прямоерефрактометрическое детектирование фракций воды(22% в ацетонитриле).
д - Прямое рефрактометрическое детектирование фракций воды(10% в ацетонитриле).
е - Прямое рефрактометрическое детектирование фракцийводы(100-процентная водопроводная вода).
105
личается от механизма удерживания на обращенно-фазной колонке, где увеличение доли органического растворителяприводит к уменьшению времени удерживания веществ практически всех классов. Следует отметить, что при использовании элюентов, содержащих 15-25% воды, вода, содержащаяся в пробах углеводов, элюируется узкими зонами с очень малым временем удерживания(практически в мёртвом объёме)и без заметного разделения на фракции.
Поскольку воду можно рассматривать как соединение существенно более полярное, чем углеводы,становится объяснимым малое время удерживания её фракций даже при использовании100
- процентного ацетонитрила (коэффициент удерживания около
0,3). Механизм удерживания воды при этом, как и в случае углеводов, связан с образованием водородных связей между ОН - группами на поверхности структурных элементов воды иNH2- группами сорбента. Судя по малому времени удерживания, энергия подобных связей невелика и существенно ниже энергии образования водородной связи (19,6 кДж/моль). Но в случае использования чистого ацетонитрила как элюента, сродство воды к подвижной фазе существенно ниже, чем в случае водно-ацетонитрильных систем, что и обуславливает её заметное удерживание на колонке. При этом становится возможной реализация многократных актов сорбция - десорбция, что, в конечном счёте, и приводит к фракционированиюводы.
Анализ рис. 9 б- д позволяет заметить, что, несмотря на изменение количества фракций воды при разных соотношениях в смеси ацетонитрил - вода, хроматографические зоныможно также,как и в случае косвенного детектирования,разделить на две груп-
106
ны. Вначале выходит отдельный пик, а затем,в зависимости от условий опыта, следующая группа линий, причём их максимально наблюдаемое количество оказывается, как это особенно прослеживается на рис. 9 е,также равно трём.
Примечательно, что сравнение хроматограмм 100-процентной дистиллированной воды (рис. 9б) и водопроводной воды (рис.9е) сразу показывает их существенное отличие. Между двумя основными группами линий в случае водопроводной воды появляется новый пик (время выхода 1,88) и, кроме того, можно говорить о появлении в хроматограмме водопроводной воды дополнительных пиков на правом склоне уширенной группы линий.
Необходимо отметить, что подобные отличия не являются просто сигналами от примесей,а отражают появление в воде новых образований, отсутствующих в чистой воде, то есть это показательнарушения структуры воды.
Таким образом, результаты, полученные методами высокоэффективной жидкостной хроматографии, свидетельствуют о том, что вода существует в квантованно-фракционном состоянии.
Совокупность полученных экспериментальных данных позволила предпринять модельные исследования построения возможных многомолекулярных стабильных ассоциатов воды, которые бы удовлетворяли в своём поведении ходу изменения наблюдаемых опытных зависимостей и отвечали бы кинетико - теоретическим представлениям.
3-7.Построениегеометрических моделей ассоциатов воды. Первичная модель из 57-ми молекул воды(рис. 5),на наш азгляд,но предшествующим предположениям казалась достаточно
обоснованной для выбора её в качестве исходной единицы (или “кванта”) в дальнейших построениях. Более того, геометрическая законченность“додекаэдрического тетраэдра”, его соответствие экспериментальным и теоретическим выводам наводила на мысль либо о его универсальности как стабильного образования, либо о наличии у него таких качественно отличных свойств, которыебьзпозволяли перейти на новые этапы построения стабильных структур.
Анализ данных,полученных тремя физико-химическими методами: рефрактометрии [47], высокоэффективной жидкостной хроматографии [49] и протонного магнитного резонанса [48], на основании которых была построена и доказана геометрическая модель основной структурной ячейки воды, свидетельствует о следующем:
1.На уровне первой стадии образования стабильного структурного элемента из57-и молекул воды (додекаэдрический тетраэдр-“квант”,рис. 5)возникает новый вид межмолекулярного взаимодействия - комплементарное образование шести водородных связей между гранями различных тетраэдров - квантов (рис.10).
2. Возникновение свойства комплементарности на уровне квантов дало возможность осуществиться комплементарному взаимодействию квантов, приведшему к5-ти и 6-ти квантовым образованиям - фракциям воды(рис.11-12).
108

Рис.10.Объёмная упаковка'‘квантов”воды.Расположение тетраэдров вершиной к наблюдателю обозначено и гранью к наблюдателю обозначено /\.Взаимодействие граней возможно только в перекрёстном положении.Пунк
тирными линиями выделены простейшие пяти (пятый за центральным) и шестиквантовые образования.
109

Рис. 11. Супертетраэдр пяти“квантов”,4 тетраэдра направлены в разные стороны от плоскостей центрального тетраэдра в виде“звезды”. Три грани, направленные к наблюдателю,расположены в одной плоскости, образуя супергрань. Наложение этих плоскостей с перекрестным положением каждой граниневозможно.
110

Рис. 12. Циклическое образование из шести “квантов”-“снежинка”. Три грани, направленные к наблюдателю, расположены в одной плоскости, образуя суперграиь. Наложение этих плоскостей с перекрёстным положением каждой грани невозможно.
Ill
Важность предложенных на основе экспериментальных данных утверждений в пп. 1и 2 требует также и рассмотрения теоретических предпосылок.
Кинетико-теоретическое обоснование появления в воде “квантов”,а также пяти- и шестиквантовых образований.
Шесть центров образования водородных связей на каждой из граней тетраэдра кванта воды можно пронумеровать в двоичной(Нили О)системе и классифицировать по типам комплементарного взаимодействия следующим образом: (см. табл. 2-3).Наличие центра образования водородной связи от атома -Н обозначено как
1,а от атома -О как0. В табл. 2 64 типа граней разбиты на группы по раному количеству атомов кислорода и,соответствен но, водорода. Систематизация типов комплементарного связывания граней тетраэдров двух квантов произведена в табл.3. Как видно из табл.3всего получается 12 типов комплементарных пар граней. Таким образом 24 типа граней связывания у тетраэдра дадут 244типов квантов(при124типах связей).
Для анализа полиассоциации квантов и определения максимально возможного количества квантов в ассоциате, как это уже указывалось выше, следует воспользоваться формулой ограничения ступенчатой ассоциации К М0=m(m+1)/2,где т - число звеньев,К - константа равновесия ступенчатой ассоциации для одной стадии, М0- общая концентрация ассоциирующих частиц [47].Напомним, что в случае определения максимального количества молекул воды в ассоциате К было выбрано 10 л/моль, а поскольку концевая молекула воды имеет три свободных центра образования водородной связи, то в расчетной формуле К получило значение 30 л/моль. Тогда количество молекул воды в квантеm=57, как это
112
Таблица 2.
Типы граней тетраэдрической модели из 57-и молекул воды./Л7
ОЮОООО
<ocwotО0*00Уоо Осс?ff
ФООiоо
оо оiо/
ооо//оаоо{//
оо/ооо(Оо/о о/оо/о'ОiyoiонО О//&°оОПь{ООi/1°
с он<(оIооо9
ОjVос}
OfоOf^о/ОVl!
сIо/ооt>/o/о/
фiO1(О
о /оН/
о//ООО(У//ПРО/
Vftо/о
О//&//
0//tСОw/f(О!Off/fOо/////
/ОООоо/осоо/
/с?е>оiО/о/^
/о &*оо/о<о/о/
{0*0ti f/сtC>O0/о/оо/
/<о/0*0
t О /ОН/0//ОО/с?//с>f/О///Оt'f!
//оооо//о&оf//oof&t/аО if
ftоfW
/fO/Of
/ f OffO / /of f f / / /ООО
1//оо/
///&/О
///.&/f////ОО////Of,////О//////
о /*
г
3
-у
<Г.6?
2
£
/<;>.//./2 -
в-/V-(£-/ъ-//.'Я_/9- 20-2/ -гг-ii- 2.V_
26 -г?.;?<?_<!?-зо~м.
зг-зз-
Зу -
*г-
3J? - 3f/ -
VC
fry ^ V3 ^
VV „ Vi^ ^6
V?-чн~
49^
Г/ -iT2-^3_УУ~-1Г_Уб—
£2--ti?-
6/ -62-63-
113
Таблица 3.
Классификацияг.::!:.'.-кошлэментгрных пар по данным табл.Р.
|
Отношение |
атокоБклслсрода |
|
Номера |
Относительная |
|
и водород: |
-i награнях |
|
граней |
вероятность |
|
п/n исходное: |
ксмелем6ятарной' |
исход; |
-:ойкомплеме |
нтарнойстыковки |
|
16/0 |
0/S |
0 |
63 |
3/64 |
|
2 3/3 |
з/з |
21 |
42 |
3/64 |
|
|
|
1 |
62 |
1/64 |
|
35/1 |
1/5 |
4 |
59 |
1/64 |
|
|
|
16 |
47 |
1/64 |
|
|
|
2 |
61 |
1/64 |
|
4 5/1 |
1 /пх/о |
В |
55 |
1/64 |
|
|
|
32 |
31 |
1/64 |
|
|
|
3 |
60 |
1/64 |
|
5 4/2 |
2/4 |
12 |
51 |
1/64 |
|
|
|
48 |
15 |
1/64 |
|
|
|
& |
58 |
1/64 |
|
6 4/2 |
2/4 |
17 |
46 |
1/64 |
|
|
|
20 |
43 |
1/64 |
|
|
|
«■> о |
57 |
1/64 |
|
7 4/2 |
2/4 |
24 |
39 |
1/64 |
|
|
|
33 |
30 |
1/64 |
|
83/3 |
3/3 |
7 28 49 |
56 35 14 |
1/64 1/64 1/64 |
|
9 4/2 |
2/4 |
о 18 36 |
54 45 2? |
1/64 1/64 1/64 |
|
10 4/2 |
2/4 |
10 34 40 |
53 29 23 |
1/64 1/64 1/64 |
11 52 1/64
11 3/3 3/3 44 19-1/64
50 13 1/64
|
|
|
22 |
41 |
1/64 |
|
12 3/3 |
3/3 |
25 |
38 |
1/64 |
|
|
37 |
26 |
1/64 |
1)4
и было показано выше, довольно точно соответствует данному ограничению 57 х 58/2=1653 и 30 х 55=1650. Применяя те же рассуждения для полиассоциации квантов какого-либо одного типа,соответственно вместе с комплементарным ему типом, концентрация которых равна 55/57 х 2/244М,и считая К=3х106л/моль по числу свободных граней концевого кванта и шестисвязному комплементарному комплексообразованию(при К=10
л/моль для образованияодной водороднойсвязи)получим,что ЗхЮ6х 55/57х2/244=m(m+1 )/2.
Отсюда m=5,4, т.е. пяти- и шестичленные образования из квантов по этому расчету могут быть ограничительными по количеству при ассоциации квантов. Поэтому целесообразно предположить наличие в водных средах преимущественного содержания термодинамически относительно устойчивых пяти- и шестиквантовых образований (фракций,наблюдаемых в хроматографическом исследовании [49]).
3.На уровне образования фракций происходит очередноекачественное преобразование межмолекулярного взаимодействия. Три шестицентровые по водородным связям грани квантов оказываются расположенными в одной плоскости (суперграни) как для фракции из 5-ти квантов (рис.11), так и для фракции из 6-ти квантов(рис. 12),приводя к возникновению нового суперкомплементарного свойства.
4. Качественным отличием суперкомплементарно-сти от простого тройного набора комплементарных граней квантов оказалась дополнительнаяизбирательность нового взаимодействия. Комплементарное взаимодействие между су-
115
пергранями одного вида фракций из-за геометрического несоответствия оказалось невозможным. Единственным вариантом комплементарного наложения в этом случае служит взаимодействие между гранями пяти- и шестиквантовых образований(рис, 13).
5. Суперкомплементарное взаимодействие между фракциями явилось последней стадией естественного усложнения свойства комплементарности стабильных ассоциатов. Это связано с тем, что при формировании из фракций структурного образования 5 + 6 + 5 (рис. 15) к тройному набору комплементарных граней квантов,расположенных в одной плоскости, неожиданно добавилась четвертая, превращая получаемый структурный элемент в простую шестигранную геометрическую фигуру(типа“елочной игрушки”,а в кристаллографических терминах - триклинная сингония)по 24 центра образования водородных связей на каждой грани, для наглядности соединенных друг с другом(рис. 16). Утверждение о последней ступени или стадии комплементарности вытекает из аналогичного для пп.1,2 кинетико-теоретическому расчету вывода о невозможности последующего комплементарного связывания каждой из таких шести граней в силу ограниченности собственной концентрации воды.
Учитывая, что в водородных связях атомы кислорода и водорода, несущие противоположные заряды, находятся на разных расстояниях от центра масс структурного элемента
116

Рис. 13. Триплет комплементарных шестицентровых связкей. Супергрань “снежинки”расположена ближе к наблюдателю (выделена более жирной линией), а супергрань “звезды”дальше от наблюдателя. Видно, что каждый треугольник грани “кванта”в “снежинке” находится в перекрёстном положении с каждым треугольником грани“кванта” в“звезде”. Кружками обозначено их пересечение, где происходит образование комплементарных связей.
117

Рис. 14. Пространственная конфигурация разных фракций воды при их взаимодействии. Соединение супертетраэдра “звезды” и цикла“снежинки” происходит по схеме, указанной • на рис. 13.
118
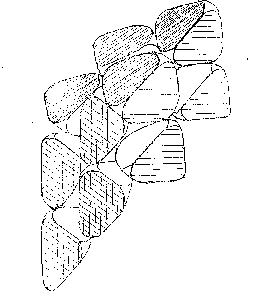
Рис. 15. Соединение одной шести и двух пятиквантовых фракций в структурный элемент воды. Разной штриховкой показано образование новых граней,включающих по 4 грани от “квантов”.
119

Рис.16. Модель структурного элемента воды. Шестигранное образование из912-ти(16 х 57)молекул воды с гранью в виде ромба с острым углом 60°. На каждой грани 24 центра образования водородной связи условно соединены между собой в шести центровые циклы.
120
(рис. 17),следует ожидать его поворота под действием электрических и магнитных полей. Ясно,что переориентации под действием физических полей подвергнутся прежде всего те элементы, которые будут максимально поляризованы или будут обладать максимальным магнитным или механическим моментами.
6. Основная структурная ячейка воды как окончательное по механизму построения стабильное структурноеоб- разование, проявляя себя в виде супермолекулы, обладает свойством лабильной слабоэнергетической кулоновской связи по граням в соответствии с распределением зарядов по 24-м центрам, т.е. своего рода “зарядоваякомплементар-пость’\Такое самокодируемое расположение структурных ячеек свидетельствует о самообусловленноетив расположении частиц и соответственно о кооперативное™их взаимодействия с находящимся в водном растворе веществом, служащим “затравкой”построения определенной ориентации частиц. Очевидно, что порядок расположения и ориентации зарядов на структурных элементах воды будет отражать соответствующее распределение электронной плотности молекул вещества “затравки”, т.е. отражать их свойства. Согласно структуре элемента это отражение будет фиксироваться всем последующим построением структурных элементов воды.
7. Утверждение об отражении свойств вещества в рисунке структурных элементов воды говорит отом, что водную среду нельзя рассматривать как относительно пассивный фактор, имеющий только термодииамико-
121
статистический или энтропийный характер. Каждая ячейка фактически оказывается информационной ячейкой и ихориентация есть не просто энергетически ориентационное поле, а своего рода энергоинформационное поле. Совокупность взаимодействующих структурных элементов воды в этом случае следует называть информационной системой воды. Следовательно, вода выступает не просто как жидкость, а как вещество, находящееся в информационно-фазовом состоянии.
Использование достаточно естественных принципов конструирования структурного элемента воды не исключает теоретического характера самого построения и потому требует каких-то более веских экспериментальных подтверждений. Подойдя к столь громадному образованию из 912-ти молекул, необходимо вспомнить, что первым указанием на близкую по количеству молекул структуру стабильного ассоциата явилось неожиданное и потому кажущееся неестественным расчётное(порядка 800)значение числа молекул в свёрнутой форме ассоциата, определённое из обработки температурной зависимости показателя преломления [47].Вторым и наиболее суще- ствнным фактором представляется несоответствие симметричной по степени электронного экранированияпротонов структуры додекаэдрического тетраэдра регистрируемому спектру ПМР. В то же время полная расшифровка наблюдаемого спектра с соответствующим соотнесением площадей пиков(см. расшифровку спектра ПМР к рис. 7)полностью отображает реальное экранирование протонов в окончательной модели
122

Рис. 17. Схематическое изображение областей положительных и отрицательных зарядов в структурномэлементе воды. Стрелками показано движение этих зарядов при повороте структурного элемента относительно центра масс.
123
структурного элемента. Третьим фактором подтверждения предлагаемой стабильной структуры ассоциата является интерпретация наблюдаемых хроматографических зон, показывающих последовательность распада структурного элемента воды. К этому выводу подводит также анализ хода зависимостей показателя преломления в смеси вода - ацетонитрил и вода - метиловый спирт, проведённый нами в работе [50]. Однако исследование взаимодействия с водой ацетонитрила и метилового спирта показало не только возможность согласования экспериментальных данных с предложенной моделью, но и позволило подойти к изучению свойств структурного элементаводы.
3-8. Бомплексообразование ацетонитрилаиметилового спирта с водой.Новые представления о возможных стабильных ассоциатах воды послужили основой для выработки качественно отличных подходов к объяснению сложных зависимостей показателя преломленияпсмесей ацетонитрил - вода и метиловый спирт - вода, приведённых на рис. 18 и 19.
Первое обстоятельство при анализе наблюдаемых кривых заключается в том, что в отличие от приведённой для сравнения на рис.18-3линейной зависимости для диметил- су л ьфокси да (ДМСО) для ацетонитрила (CH3CN) и метилового спирта (СН3ОН) наблюдаются достаточно сложные зависимости, необъяснимые в рамках обычного физического смешения жидкостей.
Для объяснения хода кривых приходится говорить о нормальном смешении Н20 и ДМСО без видимых структур-
124
CH3CN - H2O и CH3OH - H20. Поскольку из-за меньшей способности к самоассоциацииCH3CNи СН3ОН по сравнению с Н20 можно не учитывать для объяснения кривых их структурных изменений(хотя в случае СН3это влияние заметно), основные рассуждения о структурных нарушениях будут касатьсяводы.
Сложный вид зависимостей делает затруднительной их обработку, поэтому было использовано естественное предположение о том, что линейная зависимость, характерная для случая физического смешения и выполняющаяся для отдельных участков сложных кривых, свидетельствует о существовании в этих условиях двух веществ, находящихся в физической смеси. Первым линейным участком, интерпретацию которого можно было осуществить наиболее простым способом, явился участок насыщенного ацетонитрилом водного раствора. Действительно, при слабой самоассоциации ацетонитрила его разрушающее влияние на структуру воды при высоких концентрацияхдолжно былоприводить в концеконцов к полному разбитию всяких ассоциатов воды. Для проверки данного предположения намибыла проведена экстраполяция линейного участка зависимости показателя преломления счмеси ацетонитрил - вода при близких к100% концентрации ацетонитрила в воде. Если пересечение прямой при 100%CH3CNс осью ординат,дающее значение п=1,3430, естественно, совпадающее спдля ацетонитрила, представляется обычным фактом, то линейная экстраполяция данного участка к100% Н20 дало зхначениеn=1, 2830,которое сильно отличается от обычного для воды п= 1, 3334. Однако указанное значение
125

Рис. 18. Заоисимость показателя преломленияпот процентного содержания в воде ацетонитрила -1, метилового спирта - 2 и диметилсульфоксида -3.

Рис. 19. Зависимость показателя преломления пот процентного содержания в воде метилового спирта.
127
оказывается практически совпадающим с показателем преломления мономера воды, определённого из температурной зависимости п [47]. Таким образом подтверждается версия о физическом смешении компонентов на линейных участках сложной зависимости, которая в условиях, близких к 100% концентрации ацетонитрила в воде непосредственно предполагает наличие двух компонентов-ацетонитрила и мономера воды, что и показывают экспериментальные данные. Отклонение от линейности на данном участке вблизи практически уже100% - го ацетонитрила будет объяснено ниже.
Отметим, что данный вывод имеет также важное значение для интерпретации пиков в наших исследованиях методом высокоэффективной жидкостной хроматографии [49], где элю- ентом выступает ацетонитрил.
Факт полного разбития ассоциатов воды при высоких концентрациях структуроразрушающего агента заставил предположить, что и для метилового спирта при его высоком процентном содержании в воде должен наблюдаться излом в зависимости показателя преломления от соотношения спирт:вода. Данное предположение было далеко не очевидным, так как анализ хода кривой смеси СН3ОН-Н20 не давал повода искать в области 90-100% СН3ОН каких-либо резких изменений, тем более, что зависимость шла непосредственно к значению показателя преломления100%-го спирта. Однако предположение казалось обоснованным и поэтому нами, начиная со значения 90% с повышенной точностью измерения были получены ещё 10 значений до100%(рис.18и рис. 19).Результат подтвер-
128
дил ожидания: при 96% излом былобнаружен (рис. 19). Вследствие существенной самоассоциации спирта и включения мономеров воды в сетку водородных связей молекул спирта экстраполяцию кривой к мономеру воды в этом случае произвести невозможно. Эффект самоассоциации в какой-то мере присутствует и у ацетонитрила, что объясняет небольшое отклонение от линейной зависимости п вблизи100%CH3CN.
В отличие от рассмотренного участка интерпретация остальных не представляется столь очевидной и доступной вследствие наличия в водных растворах при повышении концентрации воды устойчивых ассоциатов или структурных образований из молекул воды.
Тем не менее линейный участок вблизи 100%-ой воды тоже должен быть удобным для интерпретации в отличие от участков в средней части кривых. Это связано с тем, что структурное состояние воды при небольших добавках других веществ менее всего подвержено резкому изменению. Поэтому на начальном линейном участке зависимостей показателя преломления водных смесей с ацетонитрилом и с метиловым спиртом можно предполагать наличие какой-то одной определённой структуры воды. Однако даже такое упрощающее предположение не может сразу объяснить линейностьначального участка. Дело в том, что если по версии физического смешения 2-х компонентов на линейных участках в данной области первым компонентом считать некое неизменное структурное образование из молекул воды, то вторым компонентом нельзя считать молекулу ацетонитрила или метилового спирта, поскольку в окружении избыточного количества молекул воды основное их количество всегда находится в связанном с водой состоянии. Более
129
того, речь не идёт об обычном последовательно равновесном связывании, иначе экстраполяция линейного участка приводила бы к значениям показателей преломления для CH3CNи СН3ОН (кстати для спирта п вообще лежит с другой стороны от линейного участка зависимости).Следовательно, экстраполяция приводит к значениямпоказателя преломления неких равновесно устойчивых комплексов водных структур сCH3CNи СН3ОН определённого состава. Указанный вывод может быть объяснён только существованием такой геометрии водного ассоциата, при которой возникают вполне определённые места связывания с ацетонитрилом или со спиртом.
В любом случае показатель преломления должен складываться из соответствующих долей показателя преломления водной структуры, комплекса этой структуры с ацетонитрилом (или спиртом) и, конечно, остаточного количества свободного ацетонитрила (или спирта).
Согласно результатам нашей обработки [50] при обозначениях р ,m-соответственно, количество молекул ацетонитрила и водыв комплексе удалось получить следующие соотношения: для смеси ацетонитрил-водаш/ р(р-1)=2, 677 и для смеси метиловый спирт
- вода ш/ р(р-1)=2, 673.
Фактическое совпадение значений для ацетонитрила и метилового спирта трудно объяснить лишь случайным совпадением, поскольку по своим свойствам эти вещества существенно отличаются. В этом случае естественной причиной может служить определяющая роль во взаимодействии самих структур воды, т.е. они должны обладать вполне определённым количеством мест связывания, как
130
это уже предполагалось из самого факта существования линейной зависимости.
В целых числах указанное соотношение довольно точно выражается как 8:3. Поскольку даже самая простая реализация рассматриваемого соотношения даёт числа 16/3 х 2, т.е. на 16 молекул воды пришлось бы3молекулы агента, объяснить это на основе обычных физико-химических представлений о возможных центрах связывания уже не представляется простым делом. Модель первичной стабильной структуры из 57-ми молекул воды-“кванта”позволяет найти достаточно разумное объяснение наблюдаемому необычном}’соотношению. Реализация обсуждаемого соотношения с учётом цифры 57 приводит практически к единственному решению:8/3=57 х 16/19 х 18. Поскольку 57 х 16=912 является числом молекул в предложенной модели структурного элемента воды, то необходимо рассмотреть вытекающие из модели(рис. 16)возможные места связывания с реагентами типаCH3CNи СН3ОН.
Как показано на рис. 16каждая грань модели имеет 24 центра образования водородных связей, расположенных в виде шести шестицентровых циклов -“пчелиных сот”, причём 10 центров образования водородных связей формируют 2 внутренних цикла, которые окружены 4-мявнешними циклами, состоящими из 14 центров. В отсутствие комплементарности связывания по каждой грани,что вытекает по законам комбинаторики при известной исходной концентрации молекул воды, появление мест связывания в каждой из ячеек необходимо увязывать с появлением, по крайней мере, двух одинаковых соседних зарядов в рассматриваемой ячейке (вероятность равна 1/4). Соответственно, вероятности мест посадок на 2-х внутренних циклах и 4-х внешних циклах составляют 2/6 и
131
4/6. Применяя эти значения к 10-ти внутренним и 14-ти внешним центрам образования водородных связей как к разным местам связывания, получим общую вероятность посадки, равную
1/4 х 2/6 х 60/144 +1/4 х 4/6 х 84/144=19/144
что точно соответствует единственному решению по соотношению в комплексе числа молекул реагентов и молекул воды на поверхности структурного элемента воды.
Обнаружение довольно необычного количества мест посадки на структурном элементе воды да ещё в некоторых случаях независимо от реагирующего с водой соединения подтверждает предлагаемый кулоновский характер слабого взаимодействия с ассоциа-том воды, когда определяющим для внешнего подхода оказывается соответствие распределения заряда на компонентах образующегося комплекса. Возможность для 19-ти молекул, таких как ацетонитрил или метиловый спирт, взаимодействовать с выше описанным стабильным ассоциатом воды не только характеризует рассматриваемое комплексообразование, но и оказывается своего рода базовым свойством структурного элемента воды. Дело в том, что последующее равновесное комплексообразование между элементами будет определяться комплементарностью зарядов на взаимодействующих 1ранях. С этой позиции необходимо говорить о присущем водной среде свойстве самокодируемости и кооперативности взаимодействия. Последующее равновесное создание клатратов воды размером до одного микрона становится вполне ощущаемым и реальным явлением.
Это свойство, на наш взгляд, может послужить ключом к расшифровке явления “памяти воды”, т.е. к пониманию законов по
132
строения лабильно устойчивых агрегатов из структурных элементов воды, доступных непосредственному наблюдению на контраст- но-фазовом микроскопе (рис. 20).
Изображение аналогичных лабильно устойчивых образований размером до1мкм получено также на электронном микроскопе [165,166].
3-9. Определение минимальной концентрации реагентов, определяющих информационную структуру воды.
Нами измерена концентрационная зависимость проводимости водного раствора в диапазоне сверхмалых концентраций следующих веществ: гидрофильного реагента - сахарозы (рис.21),гидрофобного реагента - бензола(рис. 22),структуроразрушающего агента - ацетонитрила(рис, 23).Обнаружены следующие эффекты:
- в области концентраций 107 -10“8М поводимость растворов остаётся фактически неизменной, выходя на плато, параллельнгое, но не равное по значению исходному плато самой воды. Остаточная“информационность”воды характеризует степень сверхслабого воздействия на структуру воды малых добавок вещества.
- наблюдаемый в области ещё более низких концентраций!О'8Минформационно-фазовый переход к исходному состоянию воды кроме ацетонитрила связан с механизмом формирования (кодирования) данным веществом информационной системы воды.
Расшифровка механизма кодирования связана с соотношением геометрии мест посадки на грани структурного элемента воды и геометрическим расположением зарядов на используемом реагенте.

Рис. 20. Изображение структуры воды, полученное на контрастно-фазовом микроскопе .Видны макрообразования размером до одного микрона, которые составлены из структурных элементов.S =1,1 х 1,1мкм,h =203 А0.
134
Рис. 21.Концентрационная.зависимость удельной проводимостиjводного раствора сахарозы. А - константа.1=Axj.
135

Рис. 22. Концентрационная зависимость удельной проводимостиjводного раствора бензола. Л - константа.I -А хj,
136
л1\
■fCi
-ю
во

Рис. 23. Концентрационная зависимость удельной проводимости jводного раствора ац его нитрил а. А - константа.I=А хj.
137
3-10. Регистрация сверхслабых электромагнитных полей информационной системой воды.
Получены кинетические зависимости изменения проводимости воды (с удельным сопротивлением18МОМ см) по сравнению с контрольным образцом после обработки воды следующими полями в течение 15-ти минут: полем магнитной мешалки(рис. 24),аэрогидрояонизатором Микулина(рис. 25),электромагнитным полем (5 гц) мотора аэрогидроионизатора Микулина на расстоянии 40 см(рис. 26).Получены также аналогичные эффекты влияния на проводимость воды непосредственнобходе воздействия: электри-чсским полем 1,5 в (рис. 27)и на рис. 28 к влиянию электрического поля 1,5 в добавлено действие сверхслабого электромагнитного поля(5 гц)от генератора с напряжённостью поля по магнитной составляющей10’9Тесла,
Во всех случаях обнаружено ступенчатое изменение со временем проводимости, отражающей структурированное состояние воды, что свидетельствует о возможности кондуктометрическойрегистрации сверхслабых воздействий на информационную системуводы.
3-11. Кинетика информационных переходов в воде.
Пусть существует некое с!руктурированное состояние водыMi(Mj- концентрация определённым образом ориентированных частиц, структурных элементов воды(рис. 29).
В результате изменения внешних фаткоров воздействия на информационную систему воды либо в результате того, что состояниеMiявлялось равновесным и рассматривалось как промежуточ
138

о)

/О£0SOмин
Рис. 24. Кинетическая зависимость изменения удельной проводимости воды jпо сравнению с контрольным образцом после а) обратимого действия поля магнитной мешалки при обработке до10мин.б)необратимого действия при 15-ти минутной обработке.
А - константа. I=А хj.

Рис. 24
139

Рис.25. Кинетическая зависимость изменения удельной проводимости водыjпо сравнению с контрольным образцом после 15-ти минутной обработки аэрогидроионизатором акад. Микулина. Л - константа.1-Axj.
140
водимостиводыjпо сравнению с контрольным образцом после 15-ти минутной обработки полем мотора аэрогидроионизатора опытного образца, находящегося на расстоянии 40см от мотора. А-константа.I —Axj.
л!ума
141

>02.С 30
Рис.27. Кинетическая зависимость изменения удельной проводимости водыjпо сравнению с контрольным образцом в ходе воздействия электрического поля 1,5 в. А - константа.I —Axj.
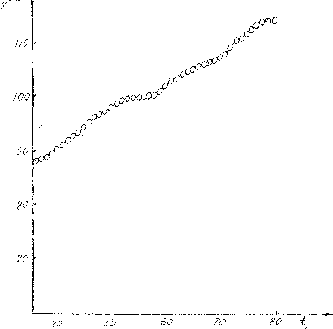
Рис. 28. Кинетическая зависимость изменения удельной проводимости воды jпо сравнению с контрольным образцом в ходе воздействия электрического поля 1,5 в и сверхслабого электромагнитногополя 5 гц от генератора с напряжённостью поля по магнитной составляющей10‘9Тесла.А-константа.I=Axj.
143
ное состояние в цепи превращений, т.е. вызывающее последующие изменения в структурном состоянии воды, в состоянииМ|должны появляться центры образования новых иначе ориентированных и организованных структур(рис. 30),прирост которых естественно будет пропорционален количеству появляющихся и по- новому организованных структур(рис.31).
Тогда измеряемый физический параметр j, пропорциональный концентрации определённого вида структур, следует записать в виде:
j = ji + (ekU-l)Cjk-jO/(ekllb -1)
где jj- параметр состоянияМ],jk- параметр полностью и необратимо изменённого состоянияМ]через времяtk, т.е. критического состояния Мк,ki.константа скорости возникновения новых образований.
Возникновение такого количества по-новому ориентированных или организованных частиц (близкого к исходной собственной концентрации) ,которое по энергии кооперативного взаимодействия превышает тепловую энергию, означает появление выделенной из предшествующего состояния структурной базы для построения устойчивого “рисунка”, т.е. нового структурного состояния М2.
Следовательно, начиная с времени tkпараметрjзаписывается уже какj=jk+(1-e'k2( 1'tk))(j2- jk)/(1-е'ща'lk))
144
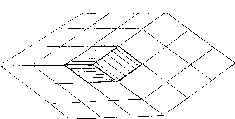
Рис.29. Структурный элемент(выделен штриховкой)среди обычной упаковки подобных себе элементов воды. Ориентация всех элементов одинаковая,
145

Рис,30. Единичное возмущение упаковки одинаково ориентированных структурных элементов в виде перевёрнутого на 180° соседнего с выделенным структурного элемента(разные штриховки).
146

Рис.31. Перестроенное под воздействием возмущения новое структурированное состояние воды. Количество пар единичной информации(разные штриховки)при энергетически выгодном их образовании способны размножаться, образуя новое состояние. Подбор пар осуществляется по принципу“зарядовой комплементарное™”граней.
147
где jk- параметр критического состоянияМк,j2-параметр состоянияМ2,к2- константа скорости перехода из критического состояния Мкв состояние М2.
Нетрудно видеть стыковку выше приведённых равенств при t=tk-Втом и другом случае получаетсяj-jk.
Очевидно,что общий подход к описанию произвольно выбранной стадии превращения структурного состояния водыМ|в№Ьпозволяет описывать любое количество последовательных превращений.
Оценка величин констант скорости перехода в соответствии с приведёнными равенствами по данным рис. 24-28 составляет (2-4)10'21/сек.
Таким образом, создаются основные принципы поведения водных систем, позволяющие объяснить проявление необычных свойств и подойти к описанию качественно нового способа перда- чи тех или иных свойств через водную среду.
Нам представляется целесообразным всё-таки изложить суть вытакающей из выше изложенного материала интересной и важной гипотезы.
3-12. Гипотеза информационнойретрапеляции.
Самокодируемость водной среды при детальном анализе кооперативного механизма взаимодействия между гранями разных структурных элементов воды приводит не только к структурированию воды. Гораздо более важным и неодинарным представляется естественная клатратность воды. Если представить, что кодирова
148
ние по определённому навязываемому рисунку заставляет структурный элемент воды закономерно объединяться друг с другом в специфические, соответствующие кодовому рисунку ограниченные по количествуэлементов образования,то мы вправе считать считать их информационными ячейками.
Используя выше рассмотренные представления об ориентационныхполях следует предположить, что взаимосвязь между ячейками, скорее всего, сохраняет исходный ориентационный пароль или код. Тогда ячеки окажутся информационно подобными и каждая из них будет как бы источником информационного влиянияи кодирования. Похоже, что мы имеем дело с явлением молекулярной информационной ретрансляции.Безусловно, что это предположение требует серьёзных доказательств в виде прямых экспериментов, однако, уже на стадии гипотезы очевидны преимущества такого способа передачи информации. Прежде всего, это независимость от расстояния и высокая надёжность. Независимость от расстояния вытекает из самого механимза зарождения ячейки от себепо доном.Действительно, если в какой-то точке водной среды формируетсязакодированная определённым образом ячейка, то через любойинтервал в отсутствие нарушения механизма информационной передачи процесс формирования очередной ячейки будет происходитьаналогично тому, как это было в исходной точке. Что касается надёжности передачи информации, то именно множественность каналов её передачи будет обеспечивать высокий информационный уровень даже при нарушении способности к регенерации ячеек в какой-либо части водной среды.
Впоследствии, вероятно, будут рассмотрены все пути трансформации передаваемого информационного рисунка, особенно при
149
наложении действия разных источников зарождения информации. Скорее всего это приведёт к рассмотрению понятия дивергенции информации, поскольку сосуществование нескольких нескольких кодообразующих рисунков в одном клатратеможет оказаться взаимозависимым. Следовательно, пересечение информационных путей может оказаться причиной некоторого изменения каждого из передаваемых информационных рисунков, т.е. будет происходить расхождение или дивергенция исходной передаваемой информации.
Гипотеза информационной ретрансляции имеет общий характер и формально может рассматриваться для любых сред. В настоящей работе её рассмотрение имеет прямой смысл, поскольку высказывается мысль о механизме информационной безопасности водной среды организма. Существование специфического для каждого организма кодового рисунка водной среды будет обеспечивать сохранноость физиологических процессов в организме только в том случае, если не произойдёт в результате внешнего воздействия такой перекодировки или наложения другого кодового рисунка, что присущий организму кодовый рисунок переориентируется в такое состояние, которое окажется неспособным поддерживать нормальное функционирование организма.
Некоторые примеры влияния на состояние водной среды в биосистемах и, соответственно, на протекание в них биохимических процессов приведены ниже.
150