

Книги по МРТ КТ на английском языке / PLUM AND POSNER S DIAGNOSIS OF STUPOR AND COM
.pdf236 Plum and Posner’s Diagnosis of Stupor and Coma
and compared psychologic changes in those patients with 10 other patients suffering from vertebral body lesions, but not cord compression, who did not receive steroids. Four of the 10 steroid-treated patients developed behavioral changes, which included hallucinations. None displayed abnormalities of alertness or state of consciousness. The control patients did not develop similar symptoms.
Depression is a more common complication in Cushing’s disease (excess pituitary adrenocorticotropic hormone [ACTH] secretion), and elation is more common after ingestion of glucocorticoids. This finding has led some investigators to hypothesize that the depressive effect of Cushing’s disease is caused by ACTH rather than cortisol. This hypothesis would be consistent with the observation that patients who have been treated with corticosteroids often become depressed as the dose is tapered and endogenous ACTH is again generated. Similar behavioral changes may be the presenting complaint in patients with paraneoplastic Cushing’s syndrome in which there is ectopic production of ACTH by a tumor (usually occult).300
Occasionally, patients with Cushing’s syndrome, particularly with adrenocorticotropinsecreting tumors, develop delirium or stupor that is not a direct result of glucocorticoid excess. Profound hypokalemic metabolic alkalosis may occur after a long period of steroid excess, and the respiratory compensation for the alkalosis may raise PaCO2 and lower PaO2, resulting in deleterious effects on the state of consciousness. Diabetes and hypertension with their attendant neurologic manifestations often complicate Cushing’s syndrome.
Thyroid Disorders
Both hyperthyroidism and hypothyroidism interfere with normal cerebral function,301,302
but exactly how the symptoms are produced is unclear. Thyroid hormone (or more strictly triiodothyronine) binds to nuclear receptors that function as ligand-dependent transcription factors. The hormone is absolutely essential for development of the brain, such that in infantile hypothyroidism the neurologic abnormality is rarely reversed unless the defect is almost immediately recognized and corrected.303 One reason may be that thyroid hormone regulates hippocampal neurogenesis in both the
juvenile and adult brain.304 Thyroid hormone also has effects on cerebral metabolism305; hypothyroidism causes a generalized decrease in regional CBF by over 20% and a 12% decrease in cerebral glucose metabolism without specific regional changes. On the other hand, hyperthyroidism appears to have little effect on cerebral metabolism.
HYPOTHYROIDISM
Coma is a rare complication of myxe- dema306–308 but one that is often associated
with a fatal outcome. In a series of 11 patients either stuporous or comatose from hypothyroidism, three of four patients who were in a coma on admission died, whereas only one of seven patients with less severe changes of consciousness died.308 Many authors have commented on the appearance of ‘‘suspended animation’’ in these profoundly hypometabolic patients. Characteristically, the patients are hypothermic with body temperatures between 878F and 918F. They appear to hypoventilate and, indeed, usually have elevated blood PCO2 values and mild hypoxia. The EEG is slow and the voltage may be either depressed or increased.309 Triphasic waves have been reported.310 The onset of myxedema coma is usually acute or subacute and precipitated by stresses such as infection, congestive heart failure, trauma, exposure to cold, or sedative or anesthetic drug administration in an untreated hypothyroid patient.
The diagnosis of myxedema in a patient in coma is suggested by cutaneous or subcutaneous stigmata of hypothyroidism, plus a low body temperature and the finding of pseudomyotonic stretch reflexes (i.e., normal jerk, but slow relaxation phase). The diagnosis is also often suggested by the presence of elevated muscle enzyme levels in the serum but can be confirmed definitively only by thyroid function tests. As myxedema coma frequently results in death, however, treatment with intravenous administration of triiodothyronine or thyroxine as well as treatment of the precipitating cause should begin once the clinical diagnosis has been made and blood for laboratory tests has been drawn; treatment should not be delayed while awaiting laboratory confirmation.
The greatest diagnostic challenge in myxedema coma is to regard one or more of its complications as the whole cause of the en-
Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
237 |
cephalopathy. Carbon dioxide narcosis may be suspected if hypoventilation and CO2 retention are present, but PaCO2 values are rarely above 50 to 55 mm Hg in hypothyroidism, and hypothermia is not part of CO2 narcosis. Some authors have attributed the cause of coma and profound hypothyroidism to respiratory failure with carbon dioxide retention, but this is unlikely as not all patients with myxedema hypoventilate. Hyponatremia is often present in severe myxedema, probably the result of inappropriate antidiuretic hormone (ADH) secretion, and sometimes is severe enough to cause seizures. Gastrointestinal bleeding and shock also can complicate severe myxedema and divert attention from hypothyroidism as a cause of coma. Hypothermia, which is probably the most dramatic sign, should always suggest hypothyroidism, but may also occur in other metabolic encephalopathies, especially hypoglycemia, depressant drug poisoning, primary hypothermia due to exposure, and brainstem infarcts.
Hashimoto’s encephalopathy is an encephalopathy associated with autoimmune thyroiditis
characterized by high titers of antithyroid antibodies in the serum.311,312 Patients may be hy-
pothyroid, but also may be euthyroid or even hyperthyroid. The disorder is a relapsing and remitting encephalopathy, and may be characterized by seizures, either focal or generalized; myoclonus; confusion; and in some instances stupor and coma. There may be associated pyramidal tract and cerebellar signs. MRI is generally uninformative; in the few cases that have come to autopsy, there is no evidence of vasculitis.313 The EEG shows generalized slowing with frontal intermittent rhythmic delta activity and often triphasic waves.314 Antithyroid antibodies are found in serum and spinal fluid, and antineuronal antibodies have also been reported in some cases, although the pathophysiologic significance of either type of antibody for the encephalopathy is not clear.313 The importance of the syndrome is that it is steroid responsive and should be suspected when a hypothyroid patient does not show an improved level of consciousness in response to thyroxin. The diagnosis is established by elevated thyroid antibodies and responsiveness to steroids.
HYPERTHYROIDISM
Thyrotoxicosis usually presents with signs of increased CNS activity (i.e., anxiety, tremor, or
hyperkinetic behavior).302,306 Subtle changes in cognitive function accompany the more obvious emotional disturbances. Rarely, in ‘‘thyroid storm,’’ these symptoms can progress to confusion, stupor, or coma.306 Thyroid storm usually develops in a patient with pre-existing thyrotoxicosis, often partially treated, who encounters precipitating factors such as an infection or a surgical procedure. The early clinical picture is dominated by signs of hypermetabolism. Fever is invariably present, profuse sweating occurs, there is marked tachycardia, and there may be signs of pulmonary edema and congestive heart failure. A more difficult problem is so-called apathetic thyrotoxicosis.315,316 Such patients are usually elderly and present with neurologic signs of depression and apathy. If untreated, the clinical symptoms progress to delirium and finally to stupor and coma. Nothing distinctive marks the neurologic picture. Hypermetabolism is not clinically prominent, nor can one observe the eye signs generally associated with thyrotoxicosis. However, almost all patients show evidence of severe weight loss and have cardiovascular symptoms, particularly atrial fibrillation and congestive heart failure. Many have signs of a moderately severe proximal myopathy. The diagnosis is established by obtaining tests that reflect thyroid hyperfunction and the neurologic signs are reversed by antithyroid treatment.
Pituitary Disorders
Pituitary failure can be associated with stupor or coma under two circumstances: (1) Pituitary apoplexy (Figure 5–8) is the term applied to hemorrhage or infarction usually of a pituitary tumor, but less commonly of the normal pituitary gland. Encephalopathy is caused by an acutely expanding mass lesion compressing the diencephalon or by inflammation due to ejection of noxious substances (blood or necrotic tissue) into the subarachnoid space. Patients generally present with headache, vomiting, photophobia, fever, visual loss, and ocular palsies. About 10% of patients are stuporous or comatose, in part due to the subarachnoid inflammation, and in part due to pituitary failure resulting from the hemorrhagic infarct.306,317 Sheehan’s syndrome, also called postpartum pituitary necrosis, is another form of pituitary
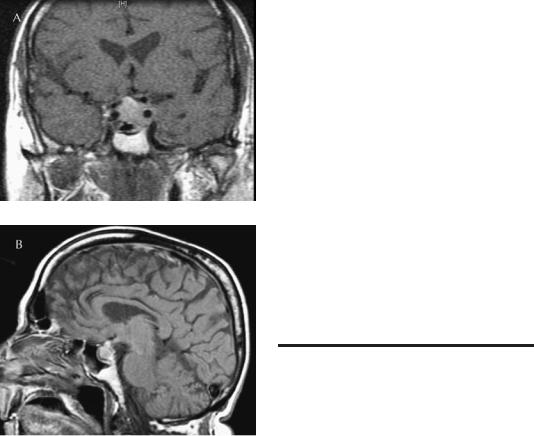
238 Plum and Posner’s Diagnosis of Stupor and Coma
Figure 5–8. Coronal (A) and sagittal (B) unenhanced T1 magnetic resonance imaging (MRI) scans of a patient with pituitary apoplexy. This 76-year-old male had been hospitalized for treatment of rectal carcinoma when he suddenly complained of headache, visual blurring, diplopia, and confusion. The MRI revealed a sellar and suprasellar mass compressing the optic chiasm and the cavernous sinuses. Surgery revealed a necrotic lesion with a few cells that probably represented a pituitary adenoma.
apoplexy. The acute form begins hours to days after delivery with signs of acute adrenal insufficiency (see above). In the past, symptoms began in the hospital before the patient went home, but because of the advent of short obstetric stays, most patients return home and then present to an emergency department with hypotension,tachycardia,hypoglycemia,fatigue, nausea, and vomiting; unrecognized, the disease is fatal.318 (2) Patients with panhypopituitarism, if the levels of corticosteroids or thyroid hormone fall low enough or if there is a disturbance of water balance, may become stuporous or comatose. In addition, similar to pa-
tients with primary adrenal or thyroid failure, patients with pain are sensitive to narcotic and sedative drugs.
Pituitary excess causes encephalopathy by hyperfunction of the pituitary-adrenal axis (i.e., during Cushing’s syndrome; see previous section).
Cancer
Diffuse encephalopathy leading to delirium, stupor, or coma is frequently seen in patients with disseminated cancer.319 About 20% of the neurologic consultations in a cancer hospital are requested for the evaluation of confused or stuporous patients.320 The causes of the mental changes are many (Table 5–11) and may
Table 5–11 Some Neurologic Complications of Cancer Causing Stupor or Coma
Lesion |
Example |
|
|
Primary brain tumor |
Hypothalamic glioma |
|
Gliomatosis cerebri |
Brain metastasis |
Carcinomatous encephalitis |
Leptomeningeal |
|
metastasis |
Hydrocephalus |
Vascular disease |
|
Large stroke |
Nonbacterial |
|
thromboendocarditis |
|
Cerebral venous |
|
occlusion |
Multiple small |
Disseminating intravascular |
strokes |
coagulation |
|
Intravascular lymphoma |
Infections |
|
Viral |
Progressive multifocal |
|
encephalopathy |
|
Herpes simplex/zoster |
Fungi |
Aspergillus |
Bacteria |
Listeria |
Side effects of |
|
therapy |
|
Radiation |
Radiation dementia |
Chemotherapy |
MTX leukoencephalopathy |
Metabolic |
Hypoglycemia |
|
Liver, renal failure |
Nutritional |
Wernicke’s |
|
Pellagra |
|
B12 deficiency |
|
encephalopathy |
|
|

Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
239 |
include all those discussed in this book.321 In a series of 140 patients with encephalopathy and cancer, two-thirds had multiple causes of their encephalopathy. However, when a single cause was identified, multiple brain metastases were the most common. In some cases, the metastases are leptomeningeal and may be discovered only by lumbar puncture. Other single causes included drugs, sepsis, multiorgan failure, and hypoxia.321 As with other patients suffering from metabolic encephalopathy, the cancer patient can often be restored to a fully sentient state if the underlying metabolic cause is corrected.
Patient 5–16
A 60-year-old man with multiple myeloma became obtunded while in the hospital. Treatment with chemotherapy had produced a severe pancytopenia, which had led to pneumonia. In addition, he suffered from renal failure and required intermittent hemodialysis. At 6:50 a.m. he was given 4 mg of levorphanol because of low back pain. Early in the afternoon he began hemodialysis, but he became hypotensive and hemodialysis was stopped. He was noted early in the evening to be markedly obtunded, with the right eye slightly deviated outward and upward. His respirations ‘‘appeared agonal.’’ On neurologic examination the patient was stuporous. With vigorous stimuli, however, he could be aroused to say his name and to identify Memorial Hospital. No other verbal responses could be secured. His pupils were 1.5 mm and reactive. In the resting position, the left eye was straight ahead and the right eye was slightly externally and superiorly deviated. Ice water calorics yielded a few beats of nystagmus in the appropriate direction. His respirations were 8 per minute, irregular, and shallow. Bilateral asterixis and extensor plantar responses were present. Laboratory abnormalities that morning had included a white blood cell count of 1,100/mm3, a hemoglobin of 9.3 g/dL, and platelets of 21,000/ mm3, and d-dimer concentrations (fibrin degradation products suggesting mild disseminated intravascular coagulation) were elevated. The serum sodium was 130 mEq/L, BUN 82 mg/dL, creatinine 5.7 mg/dL, total protein 8.1 g/L with an albumin of 3.0 g, and alkaline phosphatase of 106. Because of the small pupils and slow and shallow respiration, despite the pneumonia, the patient
was given 0.4 mg of naloxone intravenously. The pupils dilated to 6 mm, respirations went from 8 to 24 per minute, and he became awake and alert, complaining of the low back pain for which he had been given the drug that morning. The following morning he again became obtunded but less than the evening before. Pupils were 3 mm, and respirations were 20 and relatively deep. Another 0.4 mg of naloxone was given, the pupils dilated to 7 mm, respirations accelerated to 30 and deeper, and again he became alert and oriented.
Comment: The clues to opioid overdosage in this patient were the small pupils and the shallow, irregular respirations despite pneumonia. The patient’s other metabolic defects made him particularly sensitive to small doses of opioids, as did the fact that he had not received the drug in the past for pain and thus had not developed tolerance to it. Furthermore, the long action of levorphanol induced a relapse the next morning after the effects of the naloxone had worn off.
Patient 5–17
A 42-year-old woman with breast cancer known to be metastatic to bone was admitted to the hospital because of stupor. When stimulated vigorously she would answer with her name, but could not answer other questions or follow commands. On examination there was bilateral papilledema. Pupils were 2 mm bilaterally, with roving eye movements and full responses to oculocephalic maneuvers. There were diminished tendon reflexes in the left triceps and right knee jerk. Toes were upgoing. A CT scan with contrast disclosed several small enhancing lesions along the surface of the cerebral cortex. Lumbar puncture showed increased opening pressure of 300 mm/CSF, protein of 228, 14 WBCs, no RBCs, and multiple large atypical cells, which, on cytologic examination, were similar to the adenocarcinoma cells of her breast cancer. She was treated with dexamethasone and whole brain radiation therapy, resulting in rapid clearing of her cognitive function. Intraventricular chemotherapy with methotrexate and cytosine arabinoside was initiated. When she died of a pulmonary embolus 18 months later, autopsy revealed no evidence of residual cancer in the brain.
Comment: Leptomeningeal metastasis from cancer generally presents with multilevel dysfunction

240 Plum and Posner’s Diagnosis of Stupor and Coma
of the CNS, spinal cord, and spinal nerve roots. The loss of several tendon reflexes in this setting is a critical clue to the diagnosis. Radiologic evaluation may show nothing, or it may reveal superficial tumor implants along the surface of the brain, the meninges, or the spinal roots. Although often a sign of far advanced cancer, in occasional patients, particularly with breast cancer or lymphoma, vigorous treatment may clear the tumor cells and dramatically improve and extend the patient’s life.
EXOGENOUS INTOXICATIONS
Sedative and Psychotropic Drugs
Many drugs in common use can cause delirium, stupor, or coma when taken in large amounts (Table 5–12).
Table 5–12 Drugs Causing Delirium, Stupor, or Coma
Medicinal agents Amphetamines Anticholinergics Psychotropic
Tricyclics
Selective serotonin reuptake inhibitors Lithium
Phenothiazine Sedatives
Benzodiazepines Barbiturates Glutethimide Methaqualone
Opioids Acetaminophen Anticonvulsants
Nonmedical agents Alcohols
Alcohol
Ethylene glycol/propylene glycol Methanol
Illicit drugs Cocaine
Methamphetamine Gamma-hydroxybutyrate Methylenedioxymethamphetamine (MDMA) Phencyclidine
Ketamine
Rohypnol
The list of such drugs is legion; also, the agents favored by drug abusers change from time to time and differ in different geographic areas. Agents causing delirium or coma may include (1) medicinal agents prescribed but taken in overdose, (2) medicinal agents procured illicitly (e.g., opioids), (3) agents substituted for alcohol such as ethylene glycol and methanol, and (4) illicit drugs (e.g., ‘‘party’’ or ‘‘club’’ drugs).322 If it is known what agents the patient has taken, there is not much of a diagnostic problem. However, patients who are stuporous but arousable may deny drug ingestion and, if comatose, no history may be available at all.
A few drugs such as salicylates and acetaminophen can be tested at the bedside.27 Combined HPLC-immunoenzymatic screening is available in some emergency departments to detect amphetamines, barbiturates, benzodiazepines, cocaine, opioids, and phencyclidine and other drugs in 20 to 45 minutes.323 Others can be inferred from the physical examination (e.g., pupil size and response to antidotes) or rapidly procured laboratory tests. Examples include an anion gap, unidentifiable osmoles, or an oxygen saturation gap324 (Table 5–13). Measurement of the anion gap helps in establishing a diagnosis. An increased anion gap is found in toxic ingestion of drugs such as ethylene glycol, propylene glycol, methanol, paraldehyde, and salicylates. A decreased anion gap may be found after ingestion of lithium, bromides, or iodides.324 An increased osmol gap (see page 241) can be found with ethanol and ethylene ingestion. The socalled oxygen saturation gap exists when there is more than a 5% difference between calculated saturation, as measured from arterial blood, and that as measured by an oximeter. If the oximeter reading is too high after carbon monoxide intoxication, there may be severe methemoglobinemia. In addition, if the venous blood has a high oxygen content with the appearance of arterial blood, one should consider cyanide or hydrogen sulfide poisoning.324
However, in many instances, an accurate immediate diagnosis leans heavily upon the physical findings and clinical deduction. Laboratory confirmation of the clinical diagnosis is desirable, but the delay in conducting the tests often means that the information becomes available too late to be useful in guiding treatment. Furthermore, blood levels of sedatives

Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
241 |
Table 5–13 Laboratory Clues to Specific Toxins
Anion gap
Increased
Ethylene glycol
Methanol
Paraldehyde
Salicylate
Acetaminophen
Cocaine
Decreased
Bromides
Lithium
Iodide
Osmolal gap
Increased
Ethanol
Ethylene glycol
Propylene glycol
O2 saturated gap
Increased
Carbon monoxide
Methemoglobin
Cyanide
Hydrogen sulfate
Modified from Fabbri et al.323 and Mokhlesi and orbridge,324 with permission.
or alcohol sometimes provide a poor guide to the depth or anticipated duration of coma. Several reasons account for the potential discrepancy. Persons who chronically take these drugs develop a tolerance to their effects and require larger doses with resulting higher blood levels to produce coma. Pharmacologic interaction between drug mixtures and the inability to anticipate the effects of still unabsorbed material in the gut further interfere with making a correlation.
Sedatives such as benzodiazepines, neuroleptics, antihistamines, alcohol, and sedating antidepressants, as well as older drugs such as meprobamate and bromides, can all produce coma if enough is taken. The mechanism of action of each drug depends partly on its structure and partly on the dose. Many of the sedative drugs cause delirium or coma by increasing GABAergic input to the ascending arousal system, thus extinguishing wakefulness.324,326 Antidepressant drugs interfere with the reuptake of neurotransmitters, including serotonin and norepinephrine, and neuroleptics block dopamine receptors, but the more se-
dating ones also have antihistamine and anticholinergic effects. These effects may produce autonomic dysfunction, and in fact, the most dangerous effect of overdose with tricyclic antidepressants is their cardiotoxicity.
Overdoses with most depressant drugs produce fairly consistent clinical findings; individual drugs usually cause relatively minor clinical differences. Almost all of these agents depress vestibular and cerebellar function as readily as cerebral cortical function so that nystagmus, ataxia, and dysarthria accompany or even precede the first signs of impaired consciousness. Larger amounts of drug produce coma, and at this quantity all the agents depress brainstem autonomic responses. With few exceptions, such as the benzodiazepines or neuroleptics, respiration tends to be depressed at least as much as and sometimes more than somatic motor function. The pupils are usually small and reactive and ciliospinal reflexes are preserved. The oculocephalic responses are depressed or absent, and the oculovestibular responses to cold caloric testing are depressed and may be lost altogether in deep coma. Patients with depressant drug poisoning are usually flaccid with stretch reflexes that are diminished or absent. This typical picture is not always immediately seen, especially if coma develops rapidly after the ingestion of a fastacting barbiturate such as secobarbital or pentobarbital. In such cases, respiratory depression may ensue almost as rapidly as does unconsciousness; signs in the motor system may initially evolve as if function was being depressed in a rostral-caudal fashion, with a brief appearance of hyperreflexia and even clonus and extensor plantar responses. Failure to recognize this short-lived phase (it rarely lasts more than 30 to 45 minutes) as being due to depressant drugs can be fatal if one leaves the patient temporarily unattended or delays needed ventilatory assistance. The identifying clue to the toxic-metabolic basis of the changes in such cases is that the pupillary reflexes are preserved and the motor signs are symmetric. Treatment is discussed in Chapter 7.
Supportive care involves prevention of further absorption of the poison, elimination of the toxin that has already been absorbed, and, when necessary, supportive respiration, blood pressure, and cardiac rhythm. Some toxins have
specific antidotes that have been recently reviewed.324,327

Table 5–14 Clues to Specific Drugs Frequently Causing Delirium, Stupor, or Coma
|
Chemical |
|
|
Drug |
Diagnosis |
Behavior |
Physical Signs |
|
|
|
|
Amphetamine |
Blood or urine |
Hypertension; aggressive, |
Hyperthermia, hypertension, |
|
|
sometimes paranoid, |
tachycardia, arrhythmia; |
|
|
repetitive behavior |
pupils dilated; tremor, |
|
|
progressing into agitated |
dystonia, occasionally |
|
|
paranoid delirium; |
convulsions |
|
|
auditory and visual |
|
|
|
hallucinations |
|
Cocaine |
None available |
Similar to above but more |
Variable |
|
|
euphoric, less paranoid |
|
Club drugs such as |
Blood or urine |
Confused, disoriented, |
Methylenedioxy- |
|
perceptual distortions, |
methamphetamine |
|
distractible, withdrawn |
(MDMA), |
|
or eruptive; can lead to |
phenocyclidine |
|
accidents or violence |
Atropine-scopolamine |
None available |
Delirium; often agitated; |
|
|
responding to visual |
|
|
hallucinations; |
|
|
drowsiness; rarely coma |
Tricyclic |
Blood or urine |
Drowsiness; delirium; |
antidepressants |
|
agitation; rarely coma |
Phenothiazines |
Blood |
Somnolence; coma rare |
Lithium |
Blood |
Lethargic confusion, mute |
|
|
state, eventually coma. |
|
|
Multifocal seizures can |
|
|
occur. Onset can be |
|
|
delayed by hours or |
|
|
days after overdose |
Benzodiazepines |
Blood or urine |
Stupor, rarely unarousable |
Methaqualone |
Blood or urine |
Hallucinations and |
|
|
agitation blend into |
|
|
depressant drug coma |
Barbiturates |
Blood or urine |
Stupor or coma |
Alcohol |
Blood or breath |
Dysarthria, ataxia, stupor. |
|
|
Rapidly changing level |
|
|
of alertness with |
|
|
stimulation |
Opioids/opiates |
Blood or urine |
Stupor or coma |
See text
Fever, flushed face; dilated pupils; sinus or supraventricular tachycardia; hot dry skin
Fever; supraventricular tachycardia; conduction defects; ventricular tachycardia or fibrillation; hypotension; dystonia
Arrhythmias, hypotension, dystonia (see text
page 261)
Appearance of distraction; roving conjugate eye movement; pupils intact; paratonic resistance; tremors, akathisia
Essentially no cardiovascular or respiratory depression
Mild: resembles barbiturate intoxication. Severe: increased tendon reflexes, myoclonus, dystonia, convulsions. Tachycardia and heart failure
Hypothermia; skin cool and dry; pupils reactive; doll’s eyes absent; hyporeflexia; flaccid hypotension; apnea
With stupor: hypothermia, skin cold and moist;
pupils reactive, midposition to wide; tachycardia
Hypothermia; skin cool and moist; pupils symmetrically pinpoint reactive; bradycardia, hypotension; hypoventilation; pulmonary edema
242

Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
243 |
Alcoholic stupor can be a difficult diagnosis because so many patients who are unconscious for other reasons (e.g., head trauma or drug ingestion) will have the odor of ‘‘alcohol’’ (actually caused by impurities in the liquor) on their breath. Measurement of breath ethanol is not as accurate as measurement of blood ethanol and often underestimates the degree of toxicity.328 However, in a stuporous or comatose patient with a breath ethanol level of less than 50 mg/dL, alcohol intoxication is probably not the culprit and other causes need to be searched for.
The patient in an alcoholic stupor (blood level 250 to 300 mg/dL, although highly tolerant alcoholics may be awake at these levels) usually has a flushed face, a rapid pulse, a low blood pressure, and mild hypothermia, all resulting from the vasodilatory effects of alcohol. As the coma deepens (blood levels of 300 to 400 mg/dL), such patients become pale and quiet, and the pupils may dilate and become sluggishly reactive. With deeper depression respiration fails. The depth of alcoholic stupor or coma may be deceptive when judged clinically. Repetitive stimulation during medical examinations often arouses such patients to the point where they awaken and require little further stimulation to remain awake, only to lapse into a deep coma with respiratory failure when left alone in bed. Alcohol is frequently taken in conjunction with psychotropic or sedative drugs in suicide attempts. Because ethanol is also a GABAA agonist, it synergizes with the other depressant drugs. Under such circumstances of double ingestion, blood levels are no longer reliable in predicting the course, and sudden episodes of respiratory failure or cardiac arrhythmias are more frequent than in patients who have taken only a barbiturate.
HEROIN-OPIATE OVERDOSAGE
These drugs can be taken either by injection or sniffing. Overdosage with narcotics may occur from suicide attempts or, more commonly, when an addict or neophyte misjudges the amount or the quality of the heroin he or she is injecting or sniffing. Characteristic signs of opioid coma include pinpoint pupils that generally contract to a bright light and dilate rapidly if a narcotic antagonist is given. Respiratory slowing, irregularity, and cessation are prominent features and result either from di-
rect narcotic depression of the brainstem or from pulmonary edema, which is a frequent complication of heroin overdosage,329 although the pathogenesis is not understood. Opiates can cause hypothermia, but by the time such patients reach the hospital, they frequently have pneumonitis due to aspiration, so that body temperatures may be normal or elevated. Some opioids such as propoxyphene and meperidine can cause seizures. Intravenous naloxone at an initial dose of 0.2 to 0.4 mg usually reverses the effects of opioids. In patients who are physically dependent, the drug may also cause acute withdrawal. Repeated boluses at intervals of 1 to 2 hours may be needed, as naloxone is a short-acting agent and the patient may have taken a long-acting opioid.327
SEDATIVE DRUGS
The neurologic examination itself cannot categorically separate drug poisoning from other causes of metabolic brain disease. The most common diagnostic error is to mistake deep coma from sedative poisoning for the coma of brainstem infarction. The initial distinction between these two conditions may be difficult, but small, reactive pupils, absence of caloric responses, failure to respond to noxious stimuli, absence of stretch reflexes, and muscular flaccidity suggest a profound metabolic disorder. Persistent extensor responses, hyperactive stretch reflexes, spasticity, dysconjugate eye movements to caloric tests, and unreactive pupils more likely occur with brainstem destruction. If both the pupillary light reflexes and ciliospinal responses are present, deep coma is metabolic in origin. However, even if both the pupillary reactions and the ciliospinal reflexes are lost, deep coma can still be due to severe sedative intoxication. Thus, demonstration of brain death requires eliminating the possibility of a sedative overdose (see Chapter 7).
Patient 5–18
A 48-year-old woman ingested 50 g of chloral hydrate, 1.5 g of chlordiazepoxide (150 tablets of Librium), and 2.4 g of flurazepam (80 capsules of Dalmane) in a suicide attempt. Shortly afterward, her family found her in a lethargic condition and

244 Plum and Posner’s Diagnosis of Stupor and Coma
by the time they brought her to the emergency department she was deeply comatose, hypotensive, and apneic. Examination following endotracheal intubation and the initiation of artificial ventilation showed a blood pressure of 60/40 mm Hg, pupils that were 2 mm in diameter and light fixed, absent corneal and oculovestibular responses, and total muscle flaccidity accompanied by areflexia. Arterial and Schwann-Ganz catheters were placed to assist in physiologic monitoring in view of the overwhelmingly large depressant drug dose. There was already evidence of aspiration pneumonia by the time she reached the hospital. A broad-spectrum antibiotic was given and a dopamine infusion was started, which initially succeeded in raising the blood pressure to 80/60 mm Hg. By 12 hours following admission, progressively increasing amounts of dopamine to a level of 40 pg/kg/minute were unable to keep the blood pressure above 60/40 mm Hg and urine flow ceased. Treatment with L-norepinephrine was initiated at an intravenous dose that reached 12 pg/ minute. This induced a prompt rise in blood pressure to 80/40 mm Hg accompanied by a brisk urine flow. Toxicologic analysis of an admission blood sample showed the qualitative presence of chloral hydrate (quantitative assay was not available). Chlordiazepoxide level was 59.4 mg/mL and flurazepam was 6.6 mg/mL.
Early management was complicated by the effects of radiographically demonstrated aspiration pneumonia and by pulmonary edema, as well as by atrial, junctional, and ventricular premature cardiac contractions. Hypotension hovering between 80/60 and 60/40 mm Hg was a serious problem for the first 48 hours, and declines in blood pressure were repeatedly accompanied by a marginal urinary flow. The woman remained unresponsive, but by day 4 it was possible to maintain mean blood pressures above 80/60 mm Hg using dopamine; the L-norepinephrine was discontinued. Isosthenuria and polyuria developed, reflecting the probable complication of renal tubular necrosis, but meticulous attention to electrolyte balance, pulmonary toilet, and the avoidance of overhydration managed to prevent the various complications from worsening. Ice water caloric stimulation first elicited a reaction of ocular movement on day 4 and the pupillary light reflexes reappeared on the same day. On day 8 spontaneous breathing began and one could detect stretch reflexes in the extremities. She first responded to noxious stimuli by opening her eyes and withdrawing her limbs on day 10 and she
mumbled words 1 day later. Not until day 13 did she fully awaken to follow commands and answer questions. The quick phase of nystagmus to caloric stimulation did not return until day 15. She subsequently made a complete physical and intellectual recovery and received psychiatric treatment.
Comment: This woman’s course emphasizes the maxim that if patients with depressant drug poisoning survive to reach the hospital, they are potentially salvageable no matter what the blood levels of the ingested agent. The toxicologic analyses in this instance showed an amount of drug in the body that is generally regarded as a fatal dose. Whether hemodialysis would have shortened this patient’s course can be questioned, since none of the ingested agents was dialyzable. Generally speaking, among younger patients seen with drug intoxication, only those who have ingested large amounts of barbiturates have periods of unconsciousness that approach the length of this woman’s coma. However, patients put into pentobarbital coma therapeutically to treat status epilepticus may have a very similar course, and prolonged drug-induced coma does not appear to injure the brain. Her case illustrates that any sedative taken in sufficiently large amounts is capable of producing many days of coma that require meticulous systemic care to accomplish survival. Her outcome further emphasizes that even very long periods of unresponsive coma need not produce any measure of brain injury so long as blood gases and arterial perfusion pressures are maintained at levels close to the physiologic norm.
In diagnosing coma caused by depressant drug poisoning, one must not only identify the cause, but also judge the depth of coma, for the latter influences the choice of treatment. Several years ago, Reed and colleagues330 suggested a grading scheme for patients with depressant drug poisoning, as outlined in Table 5–15. The practical aspect of the classification is that only patients with grade 3 or 4 depression are at risk of losing their lives. By the same token, comparisons of the potential value of one treatment over another can only be judged by comparing them on patients in grade 3 or 4 coma, where essentially all deaths occur.
Benzodiazepines and nonbenzodiazepine agonists of the same receptors (e.g., drugs like zolpidem and eszopiclone) have replaced barbiturates as hypnotic agents. They cause much

Multifocal, Diffuse, and Metabolic Brain Diseases Causing Delirium, Stupor, or Coma |
245 |
Table 5–15 Severity of Depressant
Drug Coma*
Grade: 0 Asleep but arousable
1 Unarousable to talk but withdraws appropriately
2 Comatose; most reflexes intact; no cardiorespiratory depression
3 Comatose; no tendon reflexes; no cardiorespiratory depression
4 Respiratory failure, hypotension, pulmonary edema or arrhythmia present. Comatose for more than 36 hours
*Adapted from Reed et al.330
less respiratory depression, but at very high dosages may still cause respiratory arrest, particularly if the patient has underlying chronic pulmonary disease. An overdose can be reversed by the specific antagonist flumazenil.331 Flumazenil is useful in assessing multiagent poisoning because it reverses the side effects of the benzodiazepine; however, in some circumstances it may cause acute withdrawal seizures.332 Flumazenil does not affect coma due to alcohol, barbiturates, tricyclic antidepressants, or opioids.
INTOXICATION WITH ENDOGENOUSLY PRODUCED ‘‘BENZODIAZEPINES’’
Over the years there have been scattered case reports of patients with recurrent episodes of stupor resembling drug overdose,333 but no drug ingestion could be identified. Lugaresi and colleagues suggested the possibility that such attacks might be due to elevated levels of an endogenous benzodiazepine-like agent called ‘‘endozepine.’’334,335 Patients clinically resemble those who have taken benzodiazepines in overdose and, in fact, some have called into question whether the disorder is really due to surreptitious ingestion of benzodiazepines336; at least one of Lugaresi’s cases turned out to be due to surreptitious lorazepam ingestion.337 Stupor in such patients may last hours or days; it has an unpredictable onset and frequency. Patients are entirely normal between attacks. Like patients with benzodiazepine intoxication, these patients respond to flumazenil, which both wakes the patient and normalizes the EEG. Measures of endogenous
benzodiazepine-like levels are increased during the stupor. Patients can be treated with oral flumazenil to reduce the frequency of attacks. The first reports of the disorder may have been by Haimovic and Beresford in 1992.333
Intoxication With Other
Common Medications
Acetaminophen overdose is the most common poisoning reported to poison information centers. The drug’s metabolite (NAPQI)338 can cause acute liver necrosis, and doses above 5 g can lead to liver failure and hepatic coma. Alkalosis and grossly elevated liver function studies are a clue to its presence; prompt treatment with N-acetylcysteine often prevents fatality.339
ANTIDEPRESSANTS
These drugs include the tricyclic agents such as amitriptyline, selective serotonin reuptake inhibitors such as paroxetine and fluoxetine, and monoamine oxidase (MAO) inhibitors. All can produce delirium, and the tricyclic antidepressants can cause stupor or coma. The major toxicity of the tricyclic antidepressants is on the cardiovascular system, causing cardiac arrhythmias and hypotension. The CNS is affected by the change in blood pressure as well as the anticholinergic effects of the drugs that can lead to anhydrosis, fever, and multifocal monoclonus.327 Selective serotonin reuptake inhibitors and MAO inhibitors taken alone generally are not neurotoxic. When taken together, however, they may result in the serotonin syndrome characterized by delirium, myoclonus, hyperreflexia, diaphoresis, flushing, fever, nausea, and diarrhea. Disseminated intravascular coagulation may be a side effect and add to the CNS difficulties. Methysergide and cyproheptadine have been reported to be effective in reversing this disorder.327
Lithium intoxication is characterized by tremor, ataxia and nystagmus, choreoathetosis, photophobia, and lethargy. It may also induce nephrogenic diabetes insipidus, resulting in volume depletion and hyperosmolarity. Delirium, seizures, coma, and cardiovascular instability may occur with severe intoxication.339 Cerebellar toxicity occurs at levels higher than 3.5 mEq/L and may be nonreversible.340 With a