

|
H C |
|
|
H2C |
|
|
2 |
|
|
|
|
|
N |
CH |
2 |
N |
CH2 |
|
|
|
|
|
|
|
HO |
|
|
HO |
|
|
CH |
2 |
|
CH2 |
|
|
|
|
|
|
|
HO |
O |
O |
HO |
O |
OH |
|
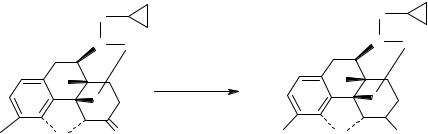
налтрексон |
|
|
6- β -налтрексол |
|
Для совместного выделения и последующего определения налтрексона и его метаболита в плазме или сыворотке крови испытуемых используют твердо-фазную экстракцию с помощью патронов Oasis MAX, фирмы «Waters». Перед анализом патроны последовательно промывают 1 мл метанола и 1 мл дистиллированной воды. Затем через патроны последовательно пропускают 1 мл плазмы, 1 мл 5 % раствора метанола в 2 % NH4OH и 1 мл 20 % раствора метанола в 2 % NH4OH. Далее пропускают 500 мкл 25 % раствора метанола в 2 % СН3СООН, который собирают в коническую колбу, нагревают 45 минут при 700С на водяной бане и аликвоту (100 мкл) вводят в хроматограф.
Для анализа используют жидкостной хроматограф с УФспектрофотометрическим детектором с переменной длиной волны. Анализ проводят при длине волны (λ ) 215 нм. Разделение проводят подвижной фазой состава ацетатный буфер с рН 5,0 (к 450 мл 0,02 М ацетата аммония добавляют уксусную кислоту до рН 5,0, затем добавляют 270 мкл триэтиламина и доводят концентрированной о-фосфорной кислотой до рН 5,0) - ацетонитрил в объемном соотношении 95:5 со скоростью подвижной фазы
1,5 мл/мин на колонке Supelco LC-18-DB 5мкм; 4,6*150мм.
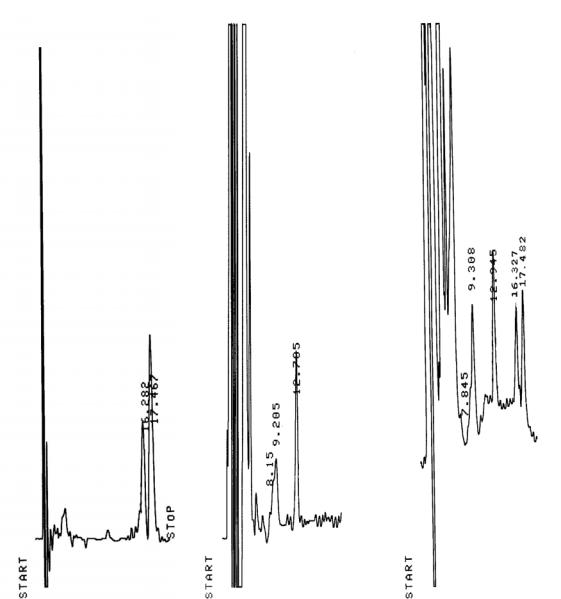
В указанных условиях время выхода пика налтрексона составляет 17,5 минут, а пика его метаболита - 16,3 минуты.
На рисунке 1 представлены хроматограммы налтрексона и его метаболита. Как видно на рисунке б место выхода пиков анализируемых соединений свободно, а появившиеся пики − рисунок в − соответствуют по времени удерживания пикам определяемых соединений − рисунок а.
Для оценки разделения исследуемых компонентов USP 24 (The United States Pharmacopeia - NF) и следующих изданий рекомендует рассчитывать параметры, характеризующие пригодность хроматографической системы:
634
Определение ламивудина
|
NH2 |
|
N |
O |
N |
CH2 |
O |
HO |
|
S |
|
Ламивудин
Биологическая жидкость. Сыворотка, плазма.
Экстракция. Выделение ламивудина из плазмы крови проводят методом твердо-фазной эктракции на патронах Sep-Pak C18 (фирмы "Waters"). Перед работой патроны промывают 1 мл метанола и 1 мл воды. Далее через патроны пропускают 1 мл плазмы крови и 1 мл воды. Затем пропускают 1 мл метанола, который собирают в конические колбы и упаривают на роторном испарителе под вакуумом при –370С. К сухому остатку добавляют 150 мкл подвижной фазы, встряхивают на vortex 1 минуту и центрифугируют 4 минуты при 10000 об/мин. Аликвоту 100 мкл вводят в хроматограф.
Хроматографический анализ. Анализ проводят на жидкостном хроматографе с УФ-спектрофотометрическим детектором с переменной длинной волны при длине волны 270 нм. В анализе используют обращеннофазную хроматографическую колонку -Bondapak 3,9*300 мм; 10 мкм (Waters). Элюирование проводят подвижной фазой состава 0,005 М КH2PO4 (рН 6,8) - метанол в соотношении 92-8. Подвижную фазу перед использованием дегазируют под вакуумом. Скорость элюирования составляетКоличественное1,0 мл/мин. определение проводят методом абсолютной калибровки.
Примечание. По аналогичной методике, немного меняя соотношение компонентов в подвижной фазе, можно определять препараты ставудин и зидовудин.
Определение индометацина
Биологическая жидкость. Сыворотка, плазма.
Экстракция. К 1 мл плазмы добавляют 200 мкл 1М о-фосфорной кислоты, встряхивают 10 секунд и добавляют 4 мл хлороформа. Смесь экстрагируют 10 минут, затем центрифугируют 10 минут при 3000 об/мин, органический слой переносят в конические колбы и упаривают на роторном испарителе под вакуумом. К сухому остатку добавляют 150 мкл подвиж-
636

|
252000 |
|
|
|
|
|
|
|
Налтрексон |
|
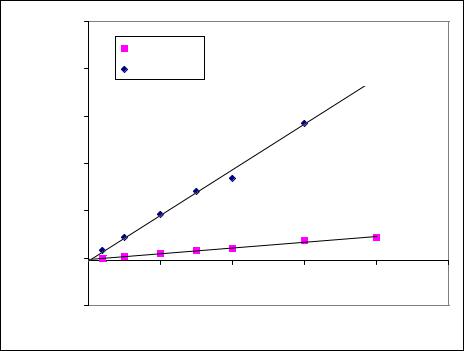
у = 1,72+0,001*х |
|
|
|
202000 |
Метаболит |
|
r2 = 0,9966 |
|
|
инт. |
152000 |
|
|
|
|
|
ед. |
|
|
|
|
|
|
|
|
|
|
|
|
|
пика, |
102000 |
|
|
у = -4,30+0,01*х |
|
|
Площадь |
|
|
|
|
||
52000 |
|
|
r2 = 0,9882 |
|
||
|
|
|
|
|
||
|
|
|
|
|
|
|
|
2000 |
|
|
|
|
|
|
0 |
50 |
100 |
150 |
200 |
250 |
|
-48000 |
|
|
|
|
|
|
|
|
Концентрация, нг/мл |
|
|
|
Рисунок 2. Калибровочный график количественного определения налтрексона и его метаболита в плазме крови
642