

акушерство
.pdfЛечение беременной состоит в проведении курсов антибактериальной терапии (ампициллин). Антибиотикотерапия показана также новорожденным, у которых при рождении диагностирован врожденный листериоз.
Урогенитальные микоплазмозы (заболевания,
вызываемые M. hominis и Ur. urealyticum). С этими инфекциями связаны невынашивание беременности, задержка роста плода, хориоамнионит, плацентит, многоводие.
Инфицирование новорожденных микоплазмами и уреаплазмами происходит редко (1- 3%). Наиболее часто возникают пневмония, менингит, острая гидроцефалия, генерализованная инфекция.
В отношении микоплазменной инфекции эффективны гентамицин и линкомицин.
Урогенитальный хламидиоз. Неблагоприятное влияние хламидиоза на плод вызывает хроническую гипоксию и задержку его роста. У новорожденных хламидиоз проявляется конъюнктивитом и пневмонией, а также фарингитом, отитом, вульвовагинитом, уретритом. Возможно развитие генерализованной инфекции и специфического миокардита, описаны случаи хламидийного менингита и энцефалита.
Проводят этиотропную антибактериальную терапию, препаратом выбора является эритромицин. Эффективны также вильпрафен, доксициклин, клиндамицин.
Гонорея. Инфицирование плода происходит преимущественно восходящим путем. Беременность может осложниться спонтанным септическим абортом, преждевременными родами, преждевременным излитием вод, хориоамнионитом, задержкой роста плода.
Гонорея проявляется у новорожденных гнойным конъюнктивитом (гонобленнорея). В раннем неонатальном периоде возможны патологическая потеря массы тела, нарушение адаптационных реакций, затянувшаяся конъюгационная желтуха, генерализованная гонококковая инфекция.
Лечение гонореи состоит в назначении антибиотиков пенициллинового ряда.
Врожденный сифилис. Заражение плода происходит внутриутробно трансплацентарно, гематогенным путем, чаще во второй половине беременности.
Ультразвуковые признаки внутриутробного сифилиса плацентомегалия, асцит и неиммунная водянка плода, задержка роста плода.
У новорожденных возможны везикулезно-буллезные высыпания на ладонях и подошвах, желтуха, геморрагический синдром, лимфаденопатия, миокардит, нефроз. Однако, как правило, признаки врожденного сифилиса проявляются значительно позже - через 3-4 нед после рождения.
Лечение матери (до 16 нед беременности) предотвращает врожденный сифилис у ребенка. Терапия в более поздние сроки беременности устраняет инфекцию у матери, но у ребенка при рождении могут наблюдаться признаки врожденного сифилиса.
Лечение беременной, больной сифилисом, проводят в соответствии с общепринятыми принципами и методами с обязательным привлечением венеролога.
Протозойные инфекции. Врожденный токсоплазмоз. Заболевание у плода может развиться только при заражении матери во время беременности. Токсоплазмоз чаще возникает у женщин, тесно контактирующих с животными (овцы, кошки), а также при употреблении сырого или недостаточно термически обработанного мяса. Клинические проявления у беременных разнообразные: увеличение лимфатических узлов, печени и селезенки, миокардит, пневмония и др.
Токсоплазмоз может сопровождаться угрозой прерывания беременности, задержкой роста плода, эндометритом. Инфекция передается трансплацентарно. Прогноз для плода зависит от сроков инфицирования беременной. В ранние сроки беременности инфицирование эмбриона нередко заканчивается его гибелью. Возможны аномалии развития: анэнцефалия, анофтальмия, микроцефалия, расщепление верхней губы, челюсти и неба (волчья пасть) и др. При заражении в более поздние сроки беременности у новорожденного отмечают триаду симптомов: гидроцефалию, хориоретинит и менингоэнцефалит с внутримозговыми петрификатами. Если заражение произошло незадолго до родов, то у плода возникают симптомы висцерального генерализованного токсоплазмоза: гепатоспленомегалия, интерстициальная пневмония, миокардит и энцефалит.
При УЗИ внутриутробное поражение фетоплацентарного комплекса при токсоплазмозе проявляется плацентомегалией, неиммунной водянкой плода, гепатоспленомегалией, внутримозговыми петрификатами, гидроцефалией.
Лечение врожденного токсоплазмоза заключаются в назначении беременной сульфаниламидных (сульфадимезин) и антипротозойных препаратов (хлоридин). В ранние сроки беременности хлоридин противопоказан в связи с опасностью тератогенного воздей-ствия на плод.
ВРОЖДЕННЫЕ АНОМАЛИИ РАЗВИТИЯ ПЛОДА
Термином "врожденные аномалии" или "врожденные пороки" обозначают любую врожденную функциональную или структурную патологию, которая выявляется у плода и новорожденного. Пороки развития могут проявляться в более поздние периоды. В зависимости от этиологии различают наследственные (генетические), экзогенные и мультифакторные врожденные аномалии развития плода.
К наследственным относятся врожденные пороки развития, возникающие вследствие генных мутаций, которые выражаются в виде эмбрионального дизморфогенеза, или хромосомных и геномных мутаций (хромосомные болезни). Происходят стойкие изменения наследственных структур в половых клетках (мутации могут быть унаследованы от одного или обоих родителей) и реже - в зиготе. В зависимости от времени воздействия тератогенного фактора врожденные пороки развития бывают следствием гаметопатий, бластопатий, эмбриопатий и фетопатий.
Под мультифакторными подразумевают пороки развития, возникающие в результате комбинированного воздействия генетических и экзогенных факторов.
Генетически обусловленные формы (генные и хромосомные) составляют около 25-30%, экзогенные (тератогенные) - 2-5%, мультифакторные -
30-40%, формы неясной этиологии - 25-50% случаев врожденных аномалий развития.
Наследственные аномалии развития плода делят на хромосомные (наиболее часто встречающиеся), моногенные и полигенные.
В основе хромосомных болезней лежат хромосомные (изменения числа или структуры хромосом) или геномные (полиплоидии) мутации. Каждому заболеванию присущи типичные кариотип и фенотип.
Практически все хромосомные аномалии (кроме сбалансированных) ведут к врожденным порокам развития. Тяжелые формы (полиплоидии, полные трисомии по аутосомам), как правило, становятся причиной самопроизвольного прерывания беременности в I триместре.
Синдром Дауна (трисомия 21) - наиболее изученная хромосомная патология, встречается с частотой 1:600 живорожденных. Цитогенетические варианты синдрома Дауна разнообразны. 94-95% случаев составляет простая полная трисомия 21 как следствие нерасхождения хромосом в мейозе (рис. 32.8). Около 2% детей с синдромом
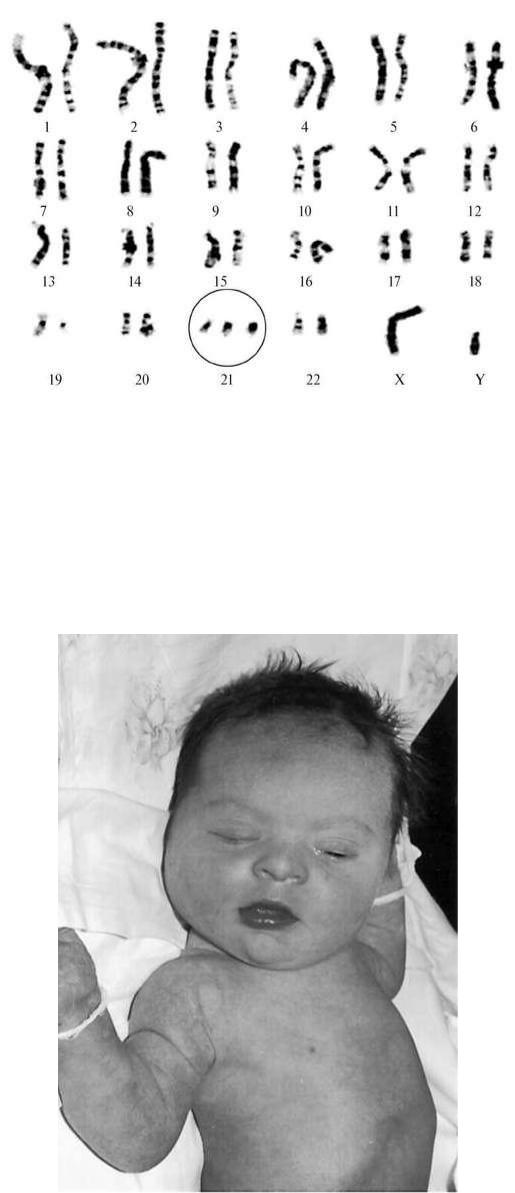
Дауна имеют мозаичные формы (47+21/46), 4% больных - транслокационную форму трисомии.
Рис. 32.8. Полная трисомия 21
Дети с синдромом Дауна имеют специфический фенотип монголоидный разрез глаз, круглое уплощенное лицо, плоскую спинку носа, эпикант, крупный (обычно высунутый) язык, брахицефалию, деформированные и низко расположенные ушные раковины, избыток кожи на шее (рис. 32.9). Часто встречаются пороки сердца, желудочнокишечного тракта, клинодактилия, четырехпальцевая (обезьянья) складка на ладони, две кожные складки вместо трех на мизинце. Отмечается задержка физического и умственного развития.
Рис. 32.9. Фенотип новорожденного с синдромом Дауна
Частота синдрома Патау (трисомия 13) составляет 1:7000 живорожденных. У 80-85% больных встречается простая полная трисомия 13 как следствие нерасхождения хромосом в мейозе у одного из родителей (чаще у матери), остальные случаи обусловлены в основном передачей дополнительной хромосомы (ее длинного плеча) в робертсоновских транслокациях типа D/13, G/13.

Синдром Патау включает в себя нарушения формирования головного мозга, глазных яблок, костей мозговой и лицевой частей черепа. Типичные признаки синдрома Патау - расщелина губы или неба, микрофтальмия, полидактилия, врожденные пороки сердца (рис. 32.10). В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни.
Рис. 32.10. Фенотип новорожденного с синдромом Патау
Синдром Эдвардса (трисомия 18) почти всегда обусловлен простой трисомной формой. Частота синдрома Эдвардса составляет 1:5000-1:7000 живорожденных. Новорожденные с синдромом Эдвардса имеют выраженную гипотрофию и множественные пороки развития лицевого черепа, сердца, костной системы, половых органов. Дети с синдромом Эдвардса, как правило, умирают в раннем возрасте.
Синдром Тернера (моносомия 45Х0) - единственная форма моносомий у живорожденных. Синдром Тернера обусловлен отсутствием одной Х-хромосомы у плодов женского пола. Частота составляет 2,5-5,5; 10 000 живорожденных женского пола. Наряду с истинной моносомией встречаются другие хромосомные аномалии по половым хромосомам (делеция короткого или длинного плеча Х-хромосомы, изохромосомы, кольцевые хромосомы, а также различные варианты мозаицизма).
Клинически синдром Тернера проявляется гипогонадизмом, врожденными пороками развития, низким ростом. Отмечаются отсутствие гонад, гипоплазия матки и маточных труб, первичная аменорея, у 25% больных встречаются пороки сердца и почек. Внешний вид больных достаточно характерен, хотя и не всегда. У новорожденных и детей грудного возраста короткая шея с избытком кожи и крыловидными складками, лимфатический отек стоп, голеней, кистей рук и предплечий (рис. 32.11). В дальнейшем проявляются отставание в росте, в развитии вторичных половых признаков, костные дисплазии, антимонголоидный разрез глаз, птоз, в 90% наблюдений - бесплодие. Часто бывает задержка психического и интеллектуального развития.

Рис. 32.11. Лимфатический отек стопы у новорожденного с синдромом Тернера
Моногенные заболевания обусловлены мутациями или отсутствием отдельного гена. Мутации могут захватывать один или оба аллеля. Клинические проявления возникают в результате отсутствия генетической информации или реализации дефектной.
Моногенные заболевания исследуются в полном соответствии с законами Менделя (аутосомное или сцепленное с
Х-хромосомой). Известно около 5000 моногенных заболеваний, более половины наследуется по аутосомно-доминантному типу.
К этой группе заболеваний относятся:
-нейрофиброматоз (болезнь Реклингхаузена), при котором наиболее тяжело поражается нервная система;
-миотоническая дистрофия с миотонией, мышечной слабостью, катарактой, сердечной аритмией, нарушенной толерантностью к глюкозе, умственной отсталостью;
-синдром Марфана - наследственная болезнь соединительной ткани. Наиболее специфическими признаками являются нарушения скелета, вывих хрусталика, сердечно-сосудистые изменения, эктазия твердой мозговой оболочки;
-синдром Элерса-Данло - врожденная гиперрастяжимость соединительной ткани в связи с нарушением синтеза коллагена, обусловленным мутациями в разных коллагеновых генах;
-фенилкетонурия, связанная с недостаточностью печеночного фермента фенилаланингидроксилазы, локус которой расположен в длинном плече хромосомы 12. Дети с фенилкетонурией рождаются здоровыми, но в первые же недели после рождения в связи с поступлением фенилаланина в организм с молоком матери развиваются клинические проявления заболевания: повышенная возбудимость, гиперрефлексия, повышенный тонус мышц, судорожные эпилептиформные припадки; от ребенка исходит "мышиный" запах. Позже развиваются умственная отсталость, микроцефалия;
-муковисцидоз (кистозный фиброз), в основе которого лежит нарушение транспорта ионов хлора и натрия через клеточные мембраны (ген муковисцидоза локализован в хромосоме 7), что приводит к избыточному выведению хлоридов. Отмечается
гиперсекреция густой слизи в клетках эндокринной части поджелудочной железы, эпителии бронхов, слизистой оболочке желудочно-кишечного тракта;
-адреногенитальный синдром (врожденная гиперплазия коры надпочечников) относится к группе наследственных нарушений синтеза стероидных гормонов. Наиболее распространенная форма врожденной гиперплазии коры надпочечников - дефицит 21-гидроксилазы, ген локализован в коротком плече хромосомы 6;
-миопатия Дюшенна, вызванная мутацией в гене, ответственном за синтез белка дистрофина (ген расположен в локусе Xq21). Заболевание проявляется прогрессирующей мышечной слабостью, дистрофией и некрозом отдельных мышечных волокон;
-гемофилия А - заболевание, сцепленное с Х-хромосомой, ген расположен в локусе Xq28, мутация гена обусловливает дефицит фактора VIII. Клинические проявления состоят в нарушении гемостаза, увеличении времени свертывания.
Полигенные болезни обусловлены взаимодействием определенных комбинаций аллелей разных локусов и экзогенных факторов. Заболевания контролируются сразу несколькими генами, не подчиняются законам Менделя и не соответствуют классическим типам аутосомно-доминантного, аутосомно-рецессивного наследования и наследования, сцепленного с Х-хромосомой. Проявление признака во многом зависит от экзогенных факторов.
Генетический риск полигенных болезней в большой степени зависит от семейной предрасположенности и от тяжести заболевания у родителей. Генетический риск полигенных болезней рассчитывают с помощью таблиц эмпирического риска.
Определить прогноз нередко сложно.
К полигенным болезным относятся врожденные пороки развития, не обусловленные хромосомной патологией. С клинической точки зрения различают изолированные (локализованные в одном органе), системные (в пределах одной системы органов) и множественные (в органах двух систем или более) врожденные пороки развития.
Наиболее часто встречаются пороки развития ЦНС.
Анэнцефалия - отсутствие полушарий мозга и свода черепа (рис. 32.12). Данная патология встречается с частотой 1:1000 новорожденных. Акрания (отсутствие свода черепа при наличии мозговой ткани) встречается гораздо реже.

Рис. 32.12. Анэнцефалия. А - эхограмма, беременность 13 недель; Б - фенотип новорожденного
Анэнцефалия часто сочетается с расщелиной губы и неба, аномалиями ушей и носа, пороками сердца, патологией желудочно-кишечного тракта и мочеполовой системы. Анэнцефалия и акрания относятся к летальным порокам развития, поэтому женщине рекомендуется прерывание беременности.
Цефалоцеле развивается в результате незакрытия нервной трубки, возникает на стадии 4 нед внутриутробной жизни и представляет собой выход мозговых оболочек через дефект костей черепа. Когда в состав грыжевого мешка входит мозговая ткань, аномалия носит название энцефалоцеле. Частота порока составляет 1:2000 живорожденных.
Цефалоцеле нередко связано с патологией хромосом (трисомия 13, 18, несбалансированные транслокации), входит в состав многих генетических синдромов.
Спинномозговая грыжа (spina bifida) - аномалия позвоночного столба в результате нарушения закрытия нервной трубки. Порок развития, при котором через дефект позвоночника выходят только оболочки спинного мозга, называют менингоцеле. Если грыжевой мешок содержит нервную ткань, то образование называется менингомиелоцеле. Поясничный и крестцовый отделы позвоночника - наиболее частая локализация дефектов. Различают spina bifida cystica (с образованием грыжевого мешка) (рис. 32.13) и spina bifida occulta, которая не сопровождается грыжевым выпячиванием. Частота варьирует в зависимости от географического региона и составляет от 0,5:1000 до 4:1000 новорожденных.
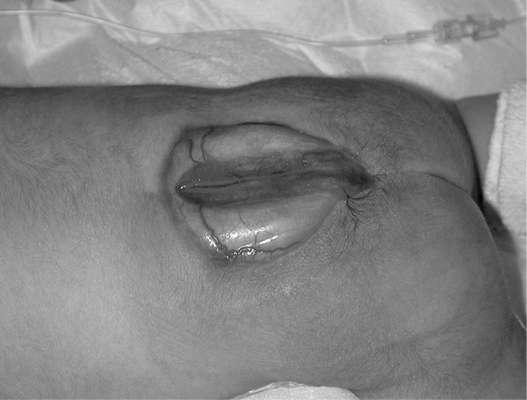
Рис. 32.13. Спинно-мозговая грыжа.А - эхограмма, беременность 17 недель; Б - новорожденный с кистозной формой спинномозговой грыжи
Дефекты нервной трубки (анэнцефалия, цефалоцеле, spina bifida) - мультифакторные аномалии, которые могут формироваться на 4-6-й неделе эмбрионального развития в результате гипертермии у матери, диабета, при воздействии на плод в ранние сроки ряда тератогенных агентов (вальпроиевая кислота, аминоптерин, метотрексат), при хромосомных аномалиях (трисомии 13, 18, триплоидия, тетраплоидии, делеции, несбалансированные транслокации), а также сочетается более чем с 40 синдромами множественных пороков развития.
При выявлении дефектов нервной трубки до достижения плодом жизнеспособности пациентке следует предложить прерывание беременности.
Для профилактики дефектов нервной трубки рекомендуют принимать фолиевую кислоту по 4 мг/сут за 3 мес до наступления беременности с последующим приемом до 6-7 нед.
Гидроцефалия - увеличение желудочков мозга с одновременным нарастанием внутричерепного давления, сопровождающееся увеличением головы. Изолированное увеличение желудочков без увеличения головы обозначают термином "вентрикуломегалия". Частота гидроцефалии 0,1:1000-2,5:1000 новорожденных.
Гидроцефалия и вентрикуломегалия, как правило, развиваются во II-III триместрах беременности в результате нарушения оттока спинномозговой жидкости, что приводит к повышению внутричерепного давления. Реже причиной гидроцефалии бывают повышенная продукция спинномозговой жидкости (папилломы сосудистых сплетений).
Гидроцефалия сопровождает многие хромосомные, моногенные заболевания, синдром множественных пороков развития, скелетных дисплазий.
При диагностике данного порока развития до жизнеспособности плода показано прерывание беременности. При отсутствии у плода хромосомной патологии и выраженных сочетанных аномалий возможно пролонгирование беременности с ультразвуковым наблюдением за нарастанием гидроцефалии.
Аномалии лицевых структур. Расщелина лица формируется между 4-й и 10-й
неделей гестации при неполном слиянии фронтоназальных структур с парными максиллярными и мандибулярными бугорками. Выделяют изолированную расщелину губы или в сочетании с расщелиной неба (наиболее частая аномалия лицевых структур) (рис. 32.14), а также изолированную расщелину неба (редкая аномалия). Расщелина может располагаться посередине, быть одноили двусторонней. Частота составляет 1:800 живорожденных.
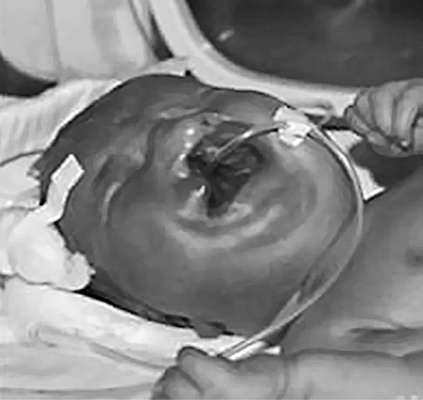
Рис. 32.14. Новорожденный с расщелиной губы и неба
Расщелины лица нередко сочетаются с другими аномалиями развития. Возникновение расщелин лица связывают с экзогенными факторами (алкоголь, фенитоин, триметадион, метотрексат), а также с инсулинзависимым сахарным диабетом у матери.
Диагностика расщелин лица возможна при УЗИ с конца I триместра беременности. Трехмерное изображение помогает уточнить диагноз. Пренатальное обследование должно включать кариотипирование и тщательное исследование (анатомия лица, мозга, сердца, скелета).
Прием фолиевой кислоты за несколько месяцев до наступления беременности снижает риск возникновения расщелин лица.
Аномалии органов грудной клетки. Врожденная диафрагмальная грыжа возникает в результате замедления закрытия плевроперитонеального канала. Дефект диафрагмы приводит к перемещению органов брюшной полости (желудок, кишечник, печень, селезенка) в грудную полость со смещением средостения и сдавлением легких (легочная гипоплазия).
Диафрагмальная грыжа нередко сочетается с пороками сердца, а также с хромосомными (трисомия 13, 18) и генными аномалиями.
Ультразвуковая диагностика возможна с конца I триместра беременности. Пренатальное обследование должно включать кариотипирование плода.
При нормальном кариотипе плода, отсутствии сочетанных аномалий беременность пролонгируют. Возможна внутриутробная хирургическая коррекция этого порока (не позднее II триместра).
Течение неонатального периода у детей с врожденной диафрагмальной грыжей зависит от выраженности гипоплазии легких и вторичной легочной гипертензии.
Врожденный кистозно-аденоматозный порок развития легких - гамартома легких,
которая представляет собой кистозное, солидное или смешанное образование в грудной клетке плода, иногда сопровождающееся водянкой плода; нередко сочетается с пороками сердца, кистозными изменениями почек, расщелиной верхнего неба и вентрикуломегалией.
Если порок представлен большим кистами, возможно внутриутробное инвазивное вмешательство - торакоамниотическое шунтирование для предупреждения гипоплазии легких. Нередко требуется хирургическая коррекция в неонатальном периоде.
Легочная секвестрация представляет собой часть легкого, которая развивается вне связи с воздухоносными путями и относится к редким порокам развития.
Секвестрированная часть легкого обычно имеет собственное кровоснабжение от сосуда, отходящего непосредственно от аорты. Наиболее часто легочная секвестрация сочетается с неиммунной водянкой.
При УЗИ легочная секвестрация визуализируется как солидное образование вблизи диафрагмы. Цветовое допплеровское картирование способствует идентификации кровоснабжения секвестрированного легкого.
Оперативное лечение ребенка после рождения заключается в сегментэктомии или лобэктомии пораженного легкого.
Пороки сердца. Частота врожденных пороков сердца у новорожденных составляет 0,5- 1%. К врожденным порокам сердца, диагностика которых в большинстве наблюдений возможна уже во внутриутробном периоде, относятся единственный желудочек, эктопия сердца, дефекты межпредсердной и межжелудочковой перегородок, гипопластический синдром левых отделов сердца, предсердно-желудочковый канал, аномалия Эбштейна, тетрада Фалло, транспозиция магистральных сосудов, артериальный ствол, стеноз и коарктация аорты, стеноз и атрезия легочной артерии, опухоли сердца. Врожденные пороки сердца нередко сочетаются с другими пороками, а также с хромосомными (трисомии) и моногенными заболеваниями.
Риск врожденных пороков сердца у плода повышен при декомпенсированном сахарном диабете, системной красной волчанке, фенилкетонурии, врожденных пороках сердца у матери. 2% всех врожденных пороков сердца связано с вирусом краснухи, алкоголем, триметадионом.
Наиболее информативным методом антенатальной диагностики врожденных пороков сердца плода является эхокардиографическое исследование, начиная со II триместра беременности. При выявлении врожденного порока сердца проводят кариотипирование плода. При сочетанных пороках и генетических аномалиях показано прерывание беременности в любом сроке. При нормальном кариотипе плода тактика ведения беременной определяется возможностью хирургической коррекции порока сердца у ребенка. Курабельность порока определяет детский кардиохирург до и после рождения ребенка.
Пороки развития желудочно-кишечного тракта. К врожденным порокам желудочно-кишечного тракта относятся атрезия пищевода, атрезия двенадцатиперстной кишки, атрезия и стеноз тонкой и толстой кишки, атрезия ануса, мекониевый перитонит.
Атрезия двенадцатиперстной кишки является наиболее частым врожденным обструктивным поражением тонкой кишки, частота данной аномалии составляет 1:10 000 живорожденных. У 30-40% плодов с атрезией двенадцатиперстной кишки диагностируются трисомия 21 и сочетанные аномалии (врожденные пороки сердца и мочевой системы, другие аномалии желудочно-кишечного тракта, дефекты позвоночника).
Диагностика порока возможна во II и III триместрах беременности. Основные ультразвуковые признаки атрезии двенадцатиперстной кишки: многоводие и классический признак "double bubble" в брюшной полости плода (рис. 32.15).
Изображение "двойного пузыря" обусловлено расширением желудка и проксимального отдела двенадцатиперстной кишки.