

3. Технические способы получения формальдегида
3.1 Каталитическое окисление метанола:
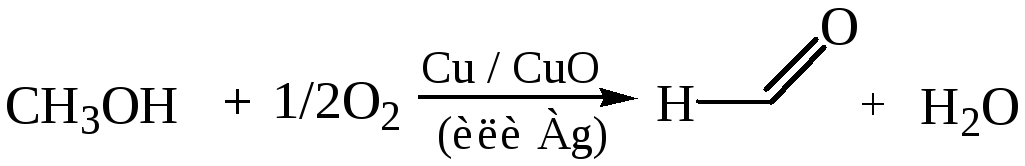
3.2 Каталитическое окисление метана:
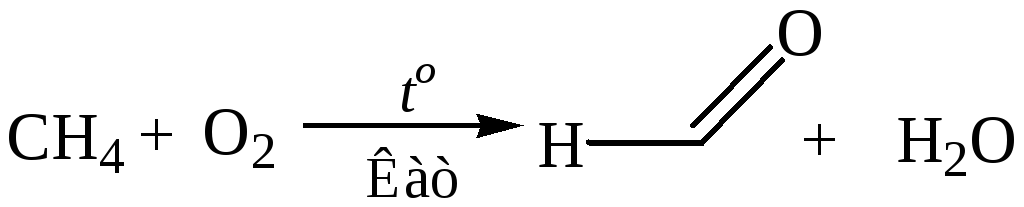
4. Восстановление производных карбоновых кислот. Для преотвращения глубокого восстановления кислоту преращают в производные, которые легче восстанавливаются, чем альдегиды, либо образуют при восстановлении соединения, легко превращающиеся в альдегиды.
4.1 Восстановление хлорангидридов кислот (по Роземунду, 1918 г.):
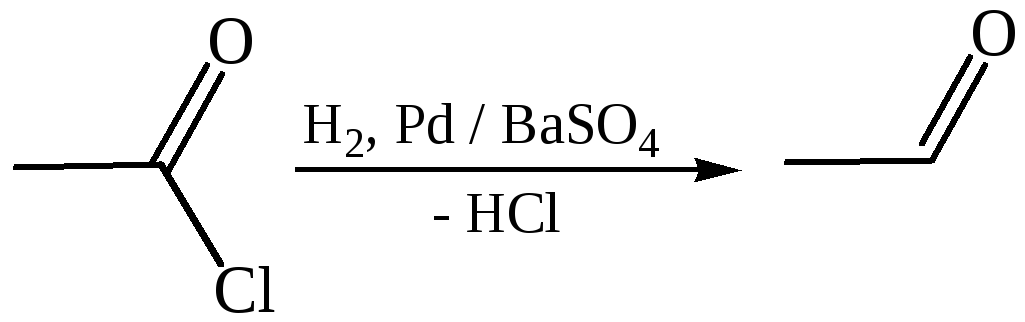
4.2 Восстановление нитрилов кислот (по Стефану, 1925 г.):
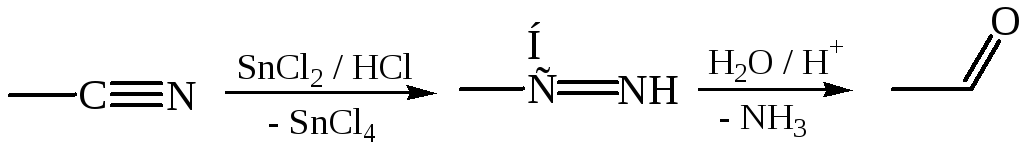
Восстановление нитрилов в альдегиды по аналогичной схеме можно осуществить LiAlH4.
5. Гидролиз дигалогенопроизводных. В результате гидролиза геминальных дигалогенопроизводных образуются гем-диолы. Гидролиз дигалогензамещенных алканов протекает по механизму нуклеофильного замещения:

6. Гидратация алкинов по Кучерову (часть 1, глава 8.4)
7. Винилирование этилового спирта ацетиленом. А.Е. Фаворский в 1887 г. открыл реакцию винилирования спиртов алкинами в присутствии щелочей:
![]()
Гидролизом винилэтилового эфира можно получить уксусный альдегид:
![]()
8. Прямое карбонилирование (оксосинтез). При обработке алкенов СО и H2 в присутствии кобальтовых катализаторов образуются альдегиды (часть 1, глава 8.2).
9. Термическое разложение солей карбоновых кислот. При нагревании кальциевых, бариевых или ториевых солей карбоновых кислот до 300 °С образуются кетоны:
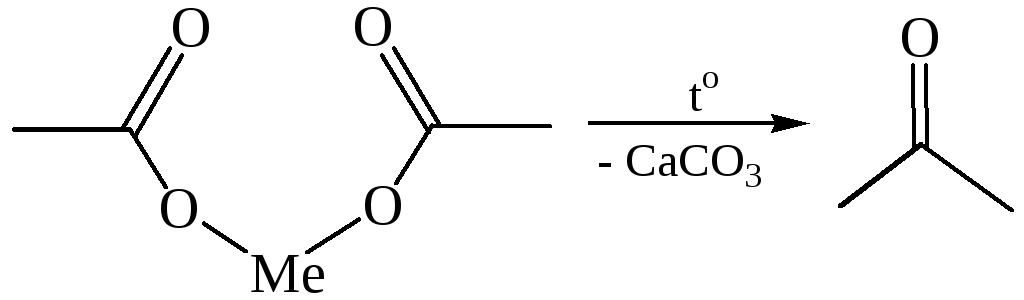
При использовании солей тория удается с удовлетворительным выходом получить макроциклические кетоны (реакция Ружички, 1926), где Сn = 7, 8, 9 и т. д. При пиролизе смеси таких солей с солями муравьиной кислоты получаются альдегиды.
10. Пинаколиновая перегруппировка (часть 2, глава 3.2)
11. Специфические методы для ароматического ряда
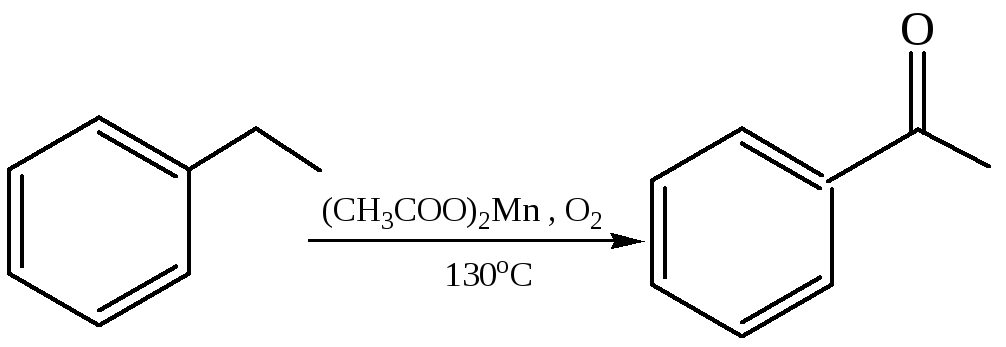
11.1 Окисление алкиларенов. Частичное окисление алкилной группы, связанной с бензольным кольцом, можно осуществить действием различных окислителей. Метильная группа – MnO2 в серной кислоте, Cr2O3 в уксусном ангидриде. В промышленности частичное окисление толуола проводят кислородом воздуха в присутствии V2O5, при этом образуется в довольно больших количествах бензойная кислота:

Окислятся кислородом в присутствии солей кобальта или марганца могут и другие боковые цепи аренов, образуя кетоны:
11.2 Ацилирование аренов (по Фриделю–Крафтсу 1877 г.). При взаимодействии аренов с хлорангидридамии кислот в присутствии кислот Льюиса в бензольном кольце атом водорода замещается на ацильный остаток, в результате образуется кетон (часть 1, глава 8.5.2). Эта реакция относится к SE реакциям бензольного кольца. Она начинается с активации реагента:
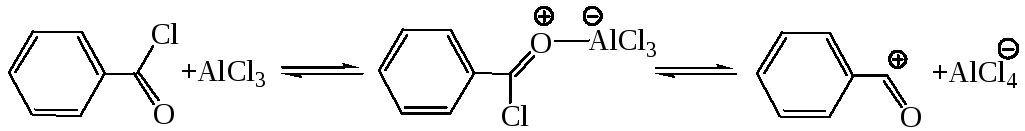
Атакующим агентом может служить либо ион ацилия, либо комплекс реагента с катализатором. Далее процесс протекает по обычной схеме SE реакции и включает стадии образования π-, затем σ-аддукта и выброс протона.
Ацилирование бензольного кольца имеет ряд особенностей, отличающих его от других SE реакций:
при ацилировании используют большое количество катализатора (не менее 1 экв.), поскольку он образует комплекс с кетоном;
ацилий-ион является слабым электрофильным реагентом. Поэтому в эту реакцию не вступают арены, содержащие сильные ЭА заместители;
ацилирующий агент имеет большой объем, поэтому у аренов с ЭД заместителями предпочтительно направление реакции – п-замещение.
11.3 Формилирование аренов (по Гаттерману–Коху 1897 г.). При обработке аренов монооксидом углерода в присутствии хлоридов алюминия и меди (I) в бензольное кольцо вводится формильная группа (часть 1, глава 8.5.2).
11.4 Получение оксиальдегидов (по Реймару–Тиману 1876 г.). Реакция основана на взаимодействии фенола с хлороформом в щелочной среде:
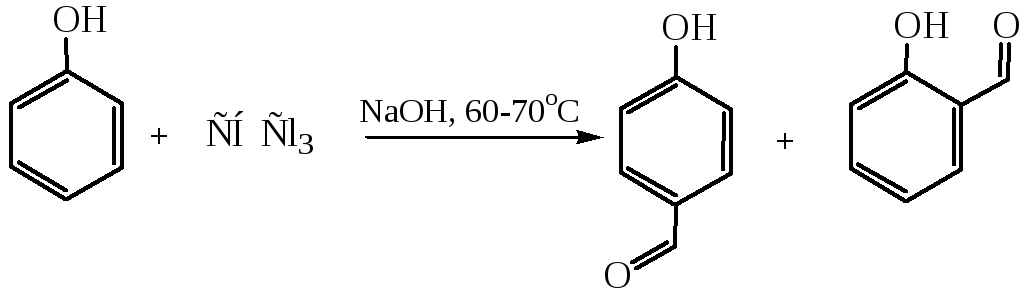
11.5 Получение оксикетонов перегруппировкой Фриса (1908 г.). При повышенных температурах в присутствии AlCl3 сложные эфиры фенолов претерпевают перегруппировку в о- и п-гидроксифенилкетоны (часть 2, глава 3.3.1).
Физические свойства и строение. За исключением газообразного формальдегида низшие альдегиды и кетоны являются жидкостями, температуры кипения которых ниже, чем у соответствующих им спиртов, так как молекулы карбонильных соединений не могут образовывать водородные связи между собой, но выше, чем у углеводородов, вследствие диполь-дипольных межмолекулярных взаимодействий (табл. 21). Способность полярных молекул альдегидов и кетонов образовывать водородные связи с водой объясняет их растворимость в воде. Удлинение углеводородного радикала увеличивает гидрофобность, что приводит к снижению растворимости в воде. Растворимость в воде и высокая растворяющая способность по отношению к неполярным и слабополярным органическим веществам делает низшие кетоны (ацетон, бутанон) удобными растворителями, но их применение в этом качестве для проведения химических реакций ограничено высокой реакционной способностью последних.
Таблица 21 – Физические свойства некоторых альдегидов и кетонов
|
Формула |
Название |
Мr |
Температура, ºС |
Растворимость в воде, г/100 г | |
|
плавления |
кипения | ||||
|
Н–СHO |
формальдегид |
30 |
–92 |
–21 |
хорошо |
|
CН3СHO |
этаналь |
44 |
–123 |
21 |
неогранич. |
|
CН3CН2СHO |
пропаналь |
58 |
–81 |
49 |
20 |
|
CН3(CН2)2СHO |
бутаналь |
72 |
–99 |
76 |
3,7 |
|
CН3СOCН3 |
пропанонон |
58 |
–95 |
56 |
неогранич. |
|
CН3СOCН2CН3 |
бутанон |
72 |
–86 |
80 |
хорошо |
Карбонильная группа является сильно полярной группой. Дипольные моменты альдегидов равны 2,5–2,6 D, а кетонов 2,7–2,8 D. В то же время карбонильная группа имеет значительную поляризуемость, т. е. на атомах карбонильной группы имеются значительные эффективные заряды, которые увеличиваются под действием внешних факторов (атакующих реагентов).
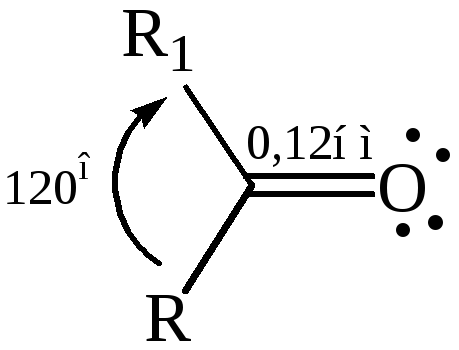
Причиной полярности и поляризуемости является особенность строения карбонильной группы. Карбонильная группа образована из двух атомов с сильно различающимися электроотрицательностями. Углеродный атом находится в sp2 гибридизации. Электронные смещения в карбонильной группе больше, чем в связи С–О, поэтому связь С=О короче по сравнению со связью С–О:
|
|
|
Природа НЭП рассматривается в двух возможных вариантах:
sp2 гибридизация и равноценность обеих электронных пар (I);
кислородный атом негибридизован, одна электронная пара находится на р-орбитали, а другая – на s-орбитали (II).
|
|
|
|
I |
II |
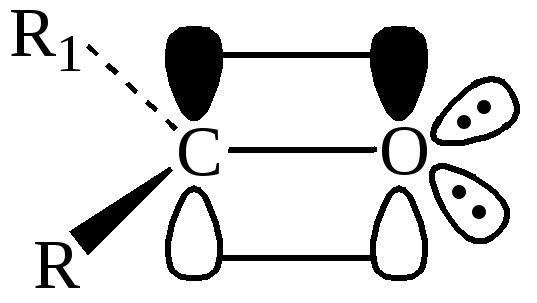
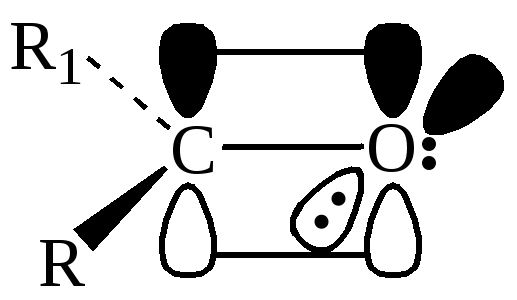
Принято считать, что атом кислорода карбонильной группы негиб-ридизован. За счет одного неспаренного р-электрона он образует с атомом углерода σ-связь. Вторая р-орбиталь, расположенная перпендикулярно плоскости σ-связей, перекрываясь с р-орбиталью углеродного атома, образует π-связь. Две оставшиеся электронные пары кислорода энергетически неравноценны: одна находится на р-орбитали, другая – на s-орбитали.
В отличие от π-МО двойной связи в алкенах в карбонильной группе энергия ВЗМО (Е1) меньше, т. е. связь С=О должна быть прочнее связи С=С. Различие между энергиями π-орбиталей ВЗМО и НСМО (Е2) у связи С=О больше, чем связи С=С, следовательно, в карбонильной группе поляризуемость π-электронного облака должна быть ниже (рисунок 1), НЭП атома кислорода занимает собственную орбиталь.
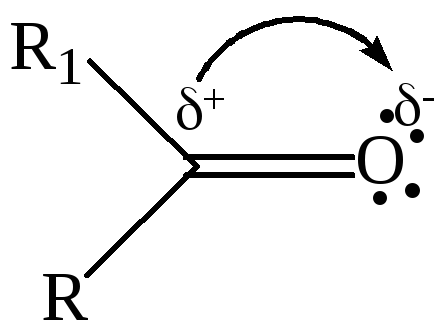
Расчеты показывают, что распределение π-электронов у связи С=О неравномерно: на углеродном атоме электронная плотность сильно понижена и смещена в сторону атома кислорода. Как следует из данных таблицы 22, двойная связь С=О осуществляется на более коротком расстоянии, она более прочная, чем двойная связь С=С, и в отличие от последней обладает большей полярностью. Высокая полярность карбонильной группы обусловлена:
различной электроотрицательностью атомов углерода и кислорода;
большей подвижностью π-электронов, которые смещены в сторону более электроотрицательного атома кислорода.
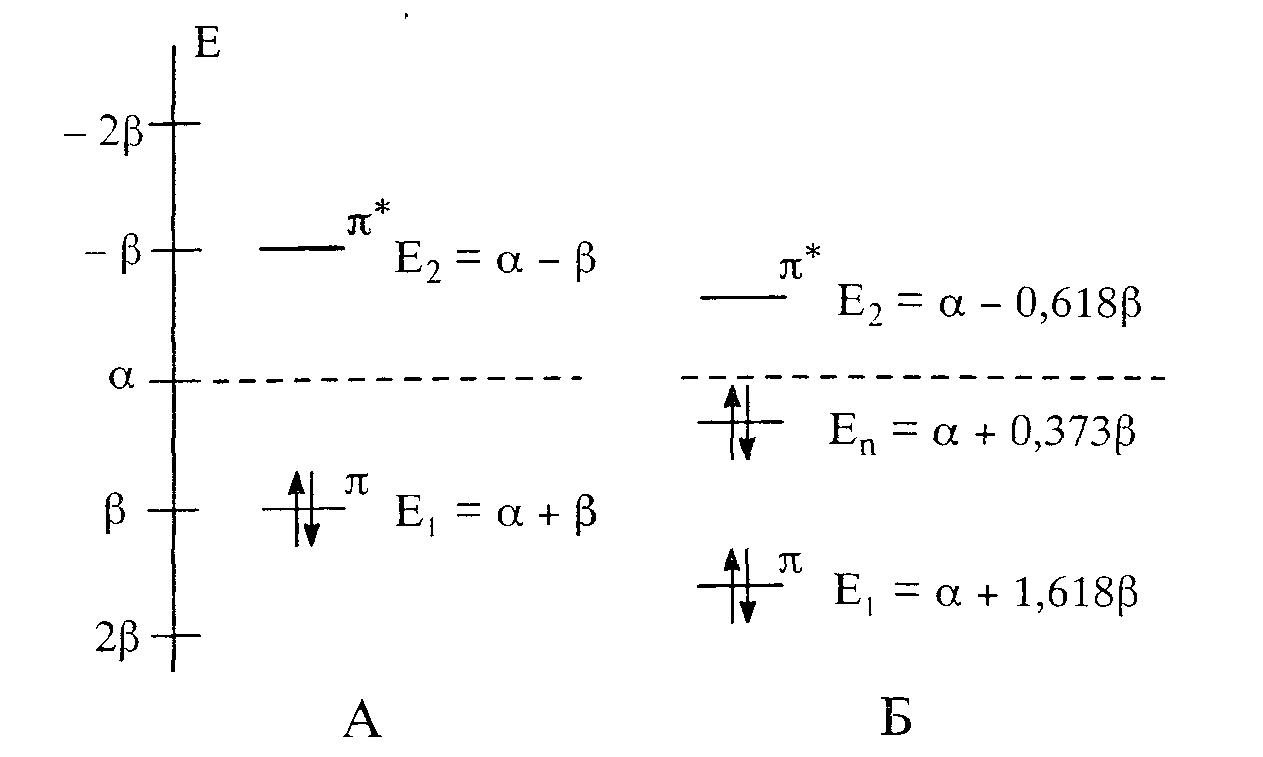
Рисунок 1 – Значения энергии π-МО:
А – для связи С=С, Б – для связиС=О
Поляризуемость связи С=О несколько ниже, чем связи С=С, однако достаточно велика. Это означает, что на углеродном атоме карбонильной группы имеется дефицит электронов, который может увеличится под действием внешних факторов, например, атакующих реагентов.
Таблица 22 – Основные физические параметры связей С=О и С=С
|
Связь |
Длина, нм |
Средняя энергия, кДж/моль |
Полярность µ, D |
Поляризуемость RD, см3 |
|
С=О |
0,121 |
690 |
2,7 |
3,3–3,5 |
|
С=С |
0,133 |
615 |
– |
4,2 |
Карбонильные соединения являются слабыми электронодонорами (сравнимы с алканолами и простыми эфирами), в то же время они обладают электроноакцепторными свойствами:

В результате присоединения электрона образуется анион-радикал, неспаренный электрон локализуется на НСМО, которая является разрыхляющей. Поэтому порядок π-связи в анион-радикале уменьшается. Часто такой анион-радикал изображается как частица с сильно локализованным неспаренным электроном:
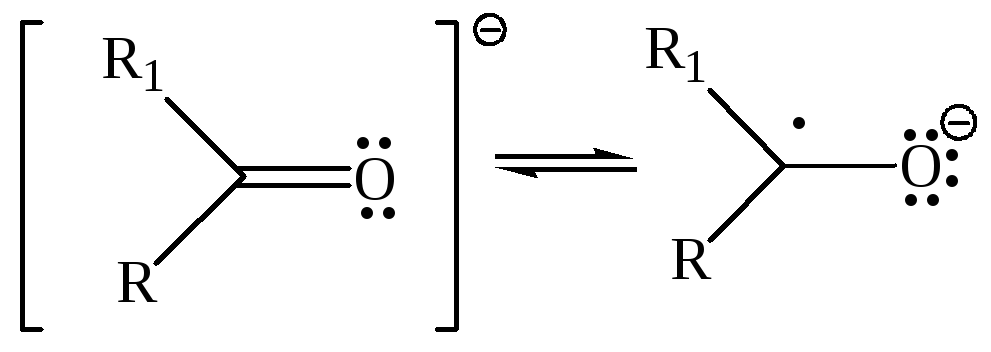
Анион-радикал карбонильных соединений образуется в реакциях с сильными восстановителями (Na, Zn).
Химические свойства. Общая характеристика реакционной способности. Углерод карбонильной группы является электронодефицитным, а кислород – электроноизбыточным центром, поэтому для карбонильной группы характерны реакции присоединения различных группировок по месту разрыва двойной связи (АN). Легкость протекания этих реакций обусловлена плоским строением группы С=О и возможностью атаки реагентов сверху или снизу от этой плоскости.
Однако вследствие значительной прочности связи С=О выгоден и обратный процесс. Поэтому большинство реакций присоединения альдегидов и кетонов является обратимыми. Скорость и положение равновесия в этих реакциях зависят от ряда факторов, таких как присутствие катализаторов, природа нуклеофила, строение субстрата и др.
Кроме АN реакций для карбонильных соединений возможны:
реакции, определяющиеся подвижностью атома водорода в алифатическом заместителе у α-углеродного атома;
реакции замещения атома водорода в ароматическом кольце, связанном с карбонильной группой;
окислительно-восстановительные реакции;
слабоосновные свойства, обусловленные НЭП на атоме кислорода.
Реакции нуклеофильного присоединения (АN)
Механизм АN реакций. АN реакции карбонильной группы протекают по следующей схеме:

Этот процесс обычно состоит из двух стадий. На первой стадии, которая является лимитирующей, происходит гетеролитический разрыв π-связи (пара электронов перемещается к кислороду), а нуклеофил за счет своей электронной пары образует σ-связь с карбонильным атомом углерода. Возникает тетраэдрический промежуточный анион, который на второй стадии процесса быстро превращается в конечный продукт присоединения (аддукт):
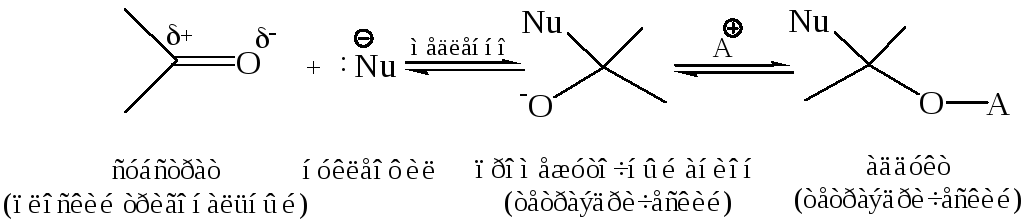
Катализ в нуклеофильных реакциях карбонильных соединений. Большинство реакций альдегидов и кетонов протекает в условиях кислотного или основного гомогенного катализа. Кислые катализаторы активируют субстрат, присоединяясь к нему посредством либо водородной связи, либо σ-связи с атомом кислорода, тем самым увеличивая дефицит электронов на углеродном атоме карбонильной группы, что можно рассматривать как кислотно-основное взаимодействие:
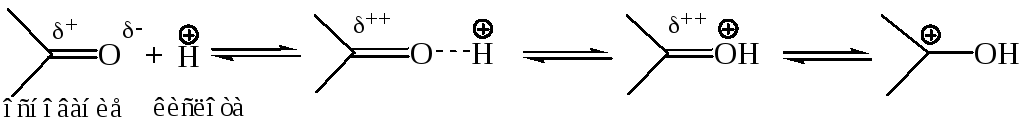
Избыток протонного катализатора отрицательно сказывается на скорости реакции, т. к. в этом случае нуклеофил может связаться с протоном в слабодиссоциирующее соединение и потерять свою активную форму. Поэтому для всех реакций карбонильных соединений, протекающих в условиях кислотного катализа, существует оптимальное значение рН, при котором скорость реакции максимальна.
В случае основного катализа роль катализатора сводится к активации реагента:
![]()
Влияние природы нуклеофила в АN реакциях. В роли нуклеофильных реагентов в реакциях альдегидов и кетонов могут выступать как нейтральные молекулы (НОН, RОН, RNН2 и др.), так и анионы (CN–, HSO3– и др.).
Природа нуклеофила определяет выбор условий проведения реакций. Сильные нуклеофилы (например, HSO3–) могут вступать в реакцию в отсутствие катализатора. Присоединение слабых нуклеофилов, как правило, требует присутствия катализатора, который активирует либо реагент, либо субстрат.
При выборе типа катализатора можно руководствоваться следующими соображениями:
если в процессе превращения возможна активация реагента, то реакцию проводят, как правило, в условиях основного катализа.
если же активировать реагент невозможно, поскольку он не обладает кислотными свойствами, то для осуществления реакции следует применить кислотные катализаторы.
Природа нуклеофильного реагента влияет также на скорость и положение равновесия АN реакций. Чем выше нуклеофильность реагента, тем выше скорость процесса, тем сильнее сдвинуто равновесие в сторону конечного продукта и более устойчив продукт превращения.
Влияние строения субстрата в нуклеофильных реакциях. На реакционную способность карбонильной группы оказывает сильное влияние характер заместителей у карбонильного атома углерода. При рассмотрении этого влияния следует принимать во внимание два фактора:
электронный;
пространственный.
При оценке влияния электронных факторов необходимо учитывать, что активность карбонильной группы зависит от величины положительного заряда (δ+) на карбонильном углероде – скорость на лимитирующей стадии тем выше, чем выше δ+. Поэтому все группировки, увеличивающие δ+ на карбонильном атоме благодаря своим ЭА свойствам, способствуют ускорению нуклеофильных реакций. И, наоборот, ЭД заместители, снижающие δ+ на реакционном центре, уменьшают скорость реакций.
При оценке влияния пространственных факторов необходимо принимать во внимание размер заместителей, связанных с карбонильной группой. Минимум энергии обеспечивается при атаке нуклеофила сверху или снизу плоскости, в которой расположена карбонильная группа (рисунок 2).
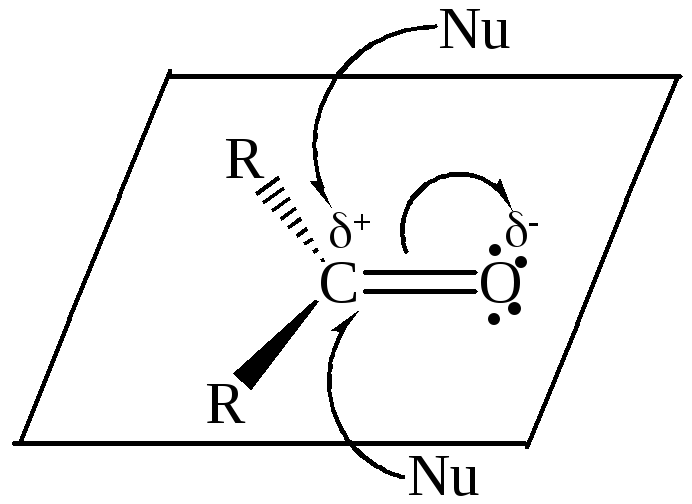
Рисунок 2 – Варианты подхода нуклеофила к карбонильной группе
При большом объеме заместителей, с одной стороны, затрудняется доступ нуклеофила к карбонильному углероду, а с другой – заместителям труднее расположиться в пространстве в тетраэдрическом аддукте.
Пространственно затрудненные кетоны не вступают в некоторые реакции присоединения, например, не взаимодействуют с большим по объему, хотя и сильным нуклеофилом NaHSO3.
Зависимость реакционной способности альдегидов и кетонов в нуклеофильных реакциях от электронных и пространственных факторов углеводородных заместителей, связанных с карбонильной группой, можно сформулировать в общем виде следующим образом:
альдегиды являются более реакционноспособными соединениями по сравнению с кетонами;
карбонильные соединения, у которых группа С=О сопряжена со связью С=С или с бензольным кольцом, менее реакционноспособны по сравнению с их насыщенными аналогами;
для замещенных бензальдегидов относительные скорости реакций уменьшаются при переходе от электроноакцепторных к электронодонорным заместителям;
пространственно затрудненные кетоны не вступают в реакции с объемными нуклеофилами.