

Квазиравновесное приближение
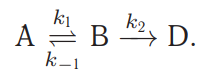
Квазиравновесное приближение применяют в том случае, когда равновесие в обратимой реакции быстро устанавливается и медленно нарушается. Для приведенной выше схемы это означает, что k2 k−1. Между собой конкурируют не процессы образования и расходования интермедиата, как при установлении квазистационарного режима, а два превращения интермедиата, одно из которых ведет к установлению равновесия, а другое — к образованию продукта реакции. Значение константы скорости k1 практически не влияет на применимость данного приближения (рис. 22.2), от него зависит только время установления квазиравновесия.
k2<<k1:
Kравн
= k1/k2
=CB/CA
→ CB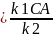 → rD=
k3CB=
→ rD=
k3CB=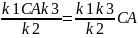
Константы скорости образования вещества Д являются комбинацией из 3-х констант отдельных стадий. Анализ применимости рассмотренных приближений показывает, что в некотором смысле они противоположны друг другу, т.е. квазистационарное приближение, применяется тогда, когда промежуточное вещество распадается быстро, а квазиравновесное, когда медленно.
НЕ НАШЛА КУСОК КАТАЛИЗА, НО БУДЕТ С РОСТОВЦЕВЫМ!!!!!!!!!
|
Простейшая схема ферментат. катализа Михаэл.-Ментена:
E+S↔(k1,k-1) [ES]→(k2) P+E
В стационарном состоянии(когда субстр. комплекса образуется столько же, сколько и расходуется): rобр[ES]= rрасп[ES] ; rобр[ES]=k1[E]равн[S] ; rраспада=k2[ES]+k-1[ES]
[E0]исходная конц. фермента=[Eравн]+[ES]
rобр=k1*([E0]-[ES])*[S] rрасп=k2*[ES]+k-1[ES]
тогда уравнение М-М: rобр=rрасп→k1([E0]-[ES])[S]=k2[ES]+k-1[ES]
[S]k1[E0]-k1[ES][S]=k2[ES]+k-1[ES] ; [S]k1[E0]-k1[ES][S]=(k2+k-1)[ES] ;
[S][E0]-[ES][S]=((k2+k-1)/k1)*[ES]
В квазистационарном состоянии (k2<<k-1) конст. скор. образ. продукта много меньше, чем константа скорости распада комплекса:k2 можно пренебречь (по условию)
k-1/k1=Kм →[ES]=([E0][S])/(Kм+[S])
Скорость образования продукта (Р) равна скорости образования фермент-субстратного комплекса [ES], а это: rобр=k2[ES], тогда получаем ур. М: rобр.р=(k2[E0][S])/(Kм+[S])-зависимость rобатной р-ии от концентрации субстрата.
→rобр.р=k2[ES]
конц. [S] при которой скорость р-ии равна ½ max.
Для большинства субстратов Км имеется в литературе, но Км кроме природы фермента и субстрата может зависеть от степени очистки, мобилизации фермента.
Физический смысл: сколько молекул субстрата подвергается превращению 1 молекулой фермента за 1 секунду. k2-число оборотов фермента.
Особенности ферментов как cat:1. Селективность (избирательность) сродство к субстрату или же способность катализировать какую то одну р-ю (один класс ферментов) в смеси;2. Оптимум рН и оптимум Т;3. Строение активного центра;4. Ферменты ускоряют р-ю в большее число раз, чем обычные cat;5. Работают в мягких условиях
|
Оксидоредуктазы: р-ии: перенос атомов водорода или е от одного в-ва к другому(дегидрогеназа, оксидаза);Трансферазы: р-ии: перенос определенной группы атомов метильной, ацильной, фосфатной или аминогруппы-одного в-ва к другому(трансаминаза, киназа);Гидролазы:р-ии: реакции гидролиза(липаза, амилаза, пептидаза);Лиазы: р-ии: негидролитическое присоединение к субстрату или отщепление от него группы атомов; при этом могут разрываться связи С-С, C-N, C-O или C-S (декарбоксилаза, фумараза, альдоза);Изомеразы:р-ии: внутримолекулярная перестройка (изомераза, мутаза);Лигазы: р-ии: соедин. 2х молекул в результате образования новых связей, сопряженное с распадом АТФ (синтетаза).
В ур Михаэлиса входит 2 константы: субстратах (Михаэлиса) и число оборотов фермента. В некоторых случаях удобнее пользоваться связанной с числом оборотов другой константой- максимальная скорость: r=(rmax[S])/(Kм+[S])
Должны знать r реакции при различных концентрациях s тогда:
(Лайнуивер-Берг) - недостатки данного способа обработки должно быть 6-7 значений; большая погрешность, так как сначала находится -1/Km а потом Km;
|
r=f(r/[S]): (Иди-Хофсти) y=kx+b, k=-Kм, x=r/[S], y=r, b=rmax, -tg=Kм; вывод: r=(rmax*[S])/(Kм+[S]) /*Kм+[S] r(Kм+[S])=rmax*[S];r*Kм+r*[S]=rmax*[S]/:[S] ((r*Kм)/[S])+r=rmax r=rmax-((r*Kм)/[S]) Достоинства: rmax и Kм находятся сразу.
Ингибирование
ферментов-прооцесс, приводящ. к замещению
р-ий. Ингибиторы делятся на несколько
гр.: 1. Конкурентные-по механизму он
блокирует активный центр фермента и
работает вместо молекул субстрата
(когда близок по св-вам к субстрату) и
тем самым снижает число доступных
центров для субстрата. Простейшая
кинетическая схема р-ии: E+S
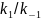 [ES]
→(k2)
P+E
фермент связывается обратимо с
ингибитором при наличии конкурентного
ингибирования: Е+I↔[EI]
; К1=[EI]/([E]*[I]).
Решить можно, если применить
квазистационарное приближение к
комплексу [ES]
и квазистационарное к комплексу фермент
с ингибитором [EI]
с учетом материального баланса по
ферменту: [E]0=[E]+[ES]+[EI]
→ r=(rmax*[S])/(Kм
эфф.+[S]),
Kм
эфф. зависит от К1
(влияния комплекса), [I]
: Kм
эфф.=Км*(1+([I]/K1),
K1
–константа
ингибирования.
[ES]
→(k2)
P+E
фермент связывается обратимо с
ингибитором при наличии конкурентного
ингибирования: Е+I↔[EI]
; К1=[EI]/([E]*[I]).
Решить можно, если применить
квазистационарное приближение к
комплексу [ES]
и квазистационарное к комплексу фермент
с ингибитором [EI]
с учетом материального баланса по
ферменту: [E]0=[E]+[ES]+[EI]
→ r=(rmax*[S])/(Kм
эфф.+[S]),
Kм
эфф. зависит от К1
(влияния комплекса), [I]
: Kм
эфф.=Км*(1+([I]/K1),
K1
–константа
ингибирования.
|
2. Неконкурентное ингибирование: E+S [ES]→P+E; Ингибитор связывается с ферментом с фермент-субстратным комплексом!
E+I↔[EI]; К1=[Ei]/[E][I] ; [ES]+I↔[ESI]; K1=[ESI]/[ES][I] → r=(rmax эфф*[S])/(Kм+[S]) ; rmax эфф=rmax/(1+([I]/K1))
Kм-не изменяется, rmax-изменяется
3. Также иногда наблюдается самый сложный тип ингибирования-смешанный.
Теории химической кинетики
Теория элементарной газофазной р-ции. Теория активных соударений.
k
=
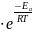 ; Еа
~ 0
; Еа
~ 0
Согласно этой теории хим. р-ция происходит при каждом столкновении реагир. частиц, соответствующим образом ориентированных относительно друг друга и обладающих энергией, равной (или большей) энергии активации. Чтобы произошла реакция, частицы в момент столкновения должны обладать некоторым минимальным избытком энергии, называемым энергией активации.
В теории активных столкновений считается, что акт превращения начальных веществ в конечные продукты совершается в момент столкновения активных молекул и протекает мгновенно. При этом молекулы рассматриваются как бесструктурные частицы, хотя в действительности химические реакции происходят путем постепенной перестройки молекул и перераспределения энергии между химическими связями.
Средняя
арифметическая скорость:
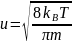 =
=
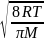
kB
= 1,38*10-23
Дж/К = 1,596*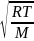
(u12)2
= (u1)2
+ (u2)2
=
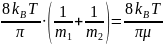 ;
;
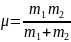 ;
;
u12
=
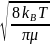 =
=
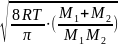 ; d12
=
; d12
=
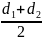
u = πd212 * u12
z (частота соударений) = πd212 * u12 * N2/u
z12
=
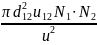 =
=
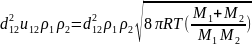 ; [м-3
* с-1]
; [м-3
* с-1]
M1
= M2
= M;
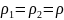
(u11)2 = (u1)2 + (u1)2 = 2(u1)2
(u11)2
=
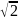 (u1)
(u1)
z12
=
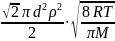 ;
;
Уравнение Трауса-Льюиса:
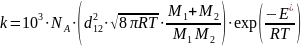
Стерический фактор.
r
= P*z*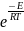
P
=
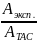 1*10-8
1*10-8
0,01 ,
а если P
,
а если P
 0,01 – медленные р-ции
0,01 – медленные р-ции
Aэксп. = Р*АTAC
Разновидности соударений.
dAB
< dXB
<
dXB – диаметр хим. взаимод.
𝜏 – 10-12 с ; Тколеб. = 10-14 с; 𝜏между соуд. = 10-10 с
Механизм «мономолекулярной» р-ции в теории активных соударений
Формальная кинетика предполагает, что порядок реакции зависит от молекулярности самой медленной стадии. Согласно теории активных столкновений химическая реакция начинается с соударения двух (значительно реже - трех) молекул. Так как большая часть реакций имеет первый порядок, то это означает, что реакция не ограничивается только столкновением двух частиц.
A
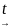 B + C
B + C
pA – парциальное давление
pA – больших r не зависит от pA0
pA – низкие n=1
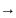 n=2
n=2Инертный газ, не участв. в р-ции в области более низких pA
Механизм реакции, объясняющий появление первого порядка при столкновении двух молекул, и называется схемой Линдемана.
Е = ½ kBT; Екин. = 3/2kT; Екин. = 3/2RT; Т=1000 К; Екин. ~ 12,5 кДж/моль
Екин << ЕА
A
+ A
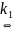 A* + A
A* + A
A*
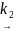 продукты
продукты
r = k2[A*]
Принцип квазистационарных концентраций:
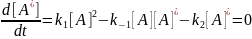
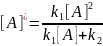 ;
;
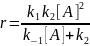
Малые pA:
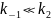
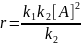 =
k1[A]2
n=2
=
k1[A]2
n=2
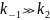
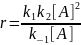 =
=
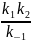 [A]
[A]
 -
эффект. константа
-
эффект. константа
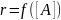
k`
=
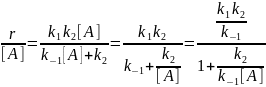
k`
=
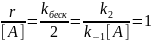
[A]
=
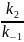
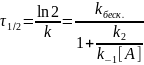
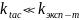
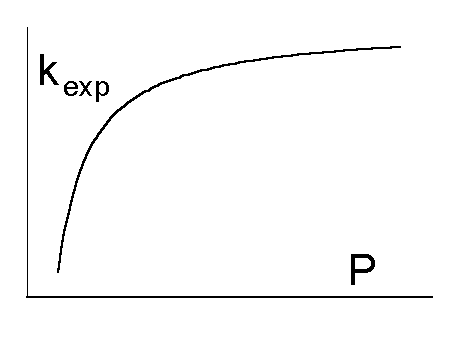
Тримолекулярные р-ции в ТАС
n=3
2NO + X2 = 2NOX;
k – слабо зависит от t°;
𝜏соуд. ≠ 0
2 стадии
Еа = 0
Н + Н + М = Н2 + М (∆Н<0 – экзотермич.)
NO + Cl2
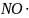 CO2
CO2
kc
=
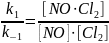
NO Cl2 + NO 2NOCl
[NO Cl2] = kc [NO][Cl2]
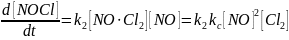
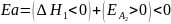
Kэксп. >> K(TAC)
Теория переходного состояния (ТПС) (активированного комплекса).
Суть теории переходного состояния (активированного комплекса):
1) частицы реагентов при взаимодействии теряют свою кинетическую энергию, которая превращается в потенциальную, и для того чтобы реакция свершилась, необходимо преодолеть некий барьер потенциальной энергии;
2) разница между потенциальной энергией частиц и упомянутым энергетическим барьером и есть энергия активации;
3) переходное состояние находится в равновесии с реагентами;
4) в тех реакциях, где энергия активации существенно ниже энергии разрыва химических связей, процессы образования новых связей и разрушения старых связей могут полностью или частично совпадать по времени.
Время существования активированного комплекса равно периоду колебания одной молекулы ( 10-13 с), поэтому он не может быть обнаружен экспериментально и, соответственно, его нельзя выделить и изучить.
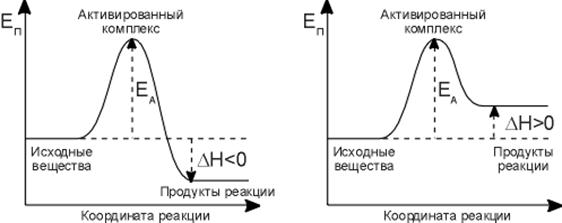
XY (исх.) + Z X + YZ (продукты)
T = 0 К
Епот. = 0 ; Еколеб. ≠ 0
Епот. = hν(u + 1/2); u = 0, 1, 2, 3…
Колеб.
число: E0
= Ea
– ½*(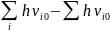 )
)
атом
+ атом: Ea
= E0
+
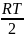
атом + лин. молекула: Ea = E0 -
2-е
линейные молекулы: Ea
= E0
-

2-е
нелинейные молекулы: Ea
= E0
-

E0 – Ea > 5 кДж/моль
Термодинамическая версия. Теория переходного состояния.
XY
(исх.) + Z
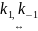 X-Y-Z
X-Y-Z
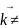 X
+ YZ
(продукты)
X
+ YZ
(продукты)
[X…Y…Z] = C≠ - конц. молекул в состоянии активир. комплекса.
C≠ << [XY] и [Z]
C≠ - быстро выполняется
k≠c
=
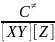 ;
k≠
-
константа скорости мономолекулярного
распада АК.
;
k≠
-
константа скорости мономолекулярного
распада АК.
C≠ - k≠c [XY][Z]
r
=
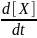 = k≠
= C≠
= { k≠
k≠c}
[XY]
[Z]
= k≠
= C≠
= { k≠
k≠c}
[XY]
[Z]
r = k[XY][Z]
k – константа скорости всей реакции
k
= k≠
*
k≠c
;
k≠c
= k≠p
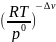 =
=
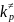 (
(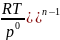
∆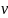 – изменение
числа молей газообр. веществ.
– изменение
числа молей газообр. веществ.
A + B C; ∆ = -1
n – порядок р-ции
∆r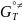 = - RT*ln
= - RT*ln
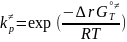
∆r
=
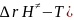
k≠c
=
(
*
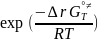 = (
*
= (
*
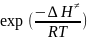 *
*
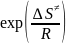
k≠
=
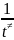 ;
;
 время
жизни активированного комплекса
время
жизни активированного комплекса
Еколеб.
= kBT
= hν
=
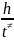 = hk≠
=
kBT
=> kB
= 1,38 * 10-23
Дж/К => k≠
=
= hk≠
=
kBT
=> kB
= 1,38 * 10-23
Дж/К => k≠
=
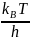 ;
;
Т = 300К; k≠ = 6,2*10-12 с-1
r(k) зависит от с≠ :
kc
=
(
*
=
(
*
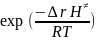 *
*
Основное уравнение т/д версии ТПС.
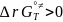 ,
C≠
<< [XY]
[Z]
,
C≠
<< [XY]
[Z]
k≠c << 1
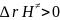 ;
k≠c
<< 1
;
k≠c
<< 1
Трансмиссионный коэффициент (ꭕ)
В уравнение ТАК следует добавить сомножитель - трансмиссионный коэффициент . Это небольшая величина, он учитывает скорость обратной реакции, которая может быть больше и меньше единицы. Значения меньшие единицы чаще всего связывают с нарушением условия адиабатичности.
XY (исх.) + Z АК Р1 Р2 Р3
Сравнение ур. т/д версии ТПС с ур. Аррениуса.
k = A*exp (-Ea/RT); А, Еа, ∆Н≠, ∆S≠ - не зависит от t°
ln
K = ln A – Ea/RT,
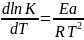
k~T*Tn-1=Tn
ln K~ln Tn = n ln T
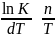
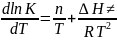 ;
;
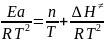 ; Еa
= nRT + ∆H≠
; Еa
= nRT + ∆H≠
Ea = ∆H≠ + 2RT; Ea = ∆H≠ + RT;
lg
K
= 15,17 -
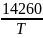 (эксп-но)
(эксп-но)
Предэкспоненциальный множитель. Энтропия активации.
k
= A*exp
(-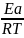 );
k
=
);
k
=
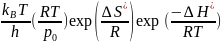 = =
= =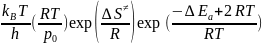
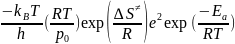
Aэксп.
= Атпс
=

Откуда
exp:
exp(-
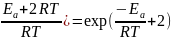
Aэксп.
= p*Атac
= p*103*NA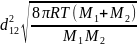
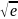
p
=
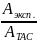 =
=
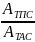 ;
АТПС
~ exp
;
АТПС
~ exp
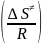
p exp
Мономолекулярные р-ции:
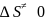
exp
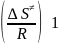
AТПС
=
(
*
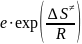 =
=
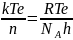
ВЫВОД: Теория активных столкновений основана на подсчете числа столкновений между реагирующими частицами, которые представляются в виде твердых сфер. Предполагается, что столкновение приведет к реакции, если выполняются два условия: 1) поступательная энергия частиц превышает энергию активации EA; 2) частицы правильно ориентированы в пространстве относительно друг друга. Первое условие вводит в выражение для константы скорости множитель exp(-EA/RT), который равен доле активных столкновений в общем числе столкновений. Второе условие дает так называемый стерический множитель P - константу, характерную для данной реакции.
В
ТАС получены два основных выражения
для константы скорости бимолекулярной
реакции. Для реакции между разными
молекулами (A + B ![]() продукты)
константа скорости равна
продукты)
константа скорости равна
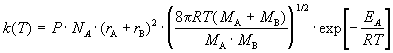
Согласно ТАС, предэкспоненциальный множитель слабо зависит от температуры. Опытная энергия активации Eоп, определяемая по уравнению (4.4), связана с аррениусовской, или истинной энергией активации EA соотношением:
Eоп = EA - RT/2.
Мономолекулярные реакции в рамках ТАС описывают с помощью схемы Линдемана (см. задачу 6.4), в которой константу скорости активации k1 рассчитывают по формулам:
и
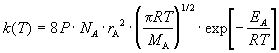
Основное уравнение теории активированного комплекса имеет вид:
|
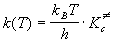
|
r=f(T, c, природы в-в и растворителя). Группы реакций: 1. Быстрые- скорость лимитируется диффузией(слабо зависят от t и мало чувствительны от природы растворителя). 2. Медленные- когда преодален энергетический барьер (Еа>40 кДж/моль) и должна сформулироваться ориентация частиц.
Молекулярные диффузии (МД). З-ны Фика.
МД-самопроизвольный перенос частиц из области с более высокой в более низкие конц., обусловлены градиентом хим. потенциала: gradμ=
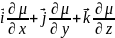
Первый з-н Фика:
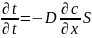 ,
D-коэф.
диффузии [м2/с];
n-кол-во
в-ва, переносимое за опред. время через
некоторую площадку S
пропорционально grad
конц. и величине поверхности через
котор. осущ. перенос.
,
D-коэф.
диффузии [м2/с];
n-кол-во
в-ва, переносимое за опред. время через
некоторую площадку S
пропорционально grad
конц. и величине поверхности через
котор. осущ. перенос.С1<C2; если идеальный р-р: μi=μ0+RT*lnCi
если S=1, то j=dn/Sdn диффузионный поток
1-ий з-н:стационарную диффузию описывает(когда постоянная величина описывается); 2-ой з-н: для нестационарной диффузии.
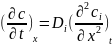 ,
T=const,
ci
–f(x,t);
зависит от скорости диффузионных
процессов; Коэф. диффузии-количеств.
хар-ка, зависит от природы в-ва и фазового
состояния.
,
T=const,
ci
–f(x,t);
зависит от скорости диффузионных
процессов; Коэф. диффузии-количеств.
хар-ка, зависит от природы в-ва и фазового
состояния.Особенности строения кристаллов и жидкостей. Клеточный эффект.
газы: Ехим→Евзаимод.(взаимод. молекул); ж-тв(крист.): ближний и дальний порядок.
Еколеб=kBT
Если Т<Tплавл. частицы сущ. в локализованном состоянии и частицы мало подвижны.
кр.→ж: физ. сва-ва меняются незначительно; (при плавлении кристалла возникает резкое повышение конц. вакансии→ж приобретает св-ва текучести→ ж обладает изотропными(одинаковыми) свойствами, которые не зависят от направления), у ж сохраняется только ближний порядок расположения частиц.
ф-ла Стокса: DA=
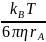 [см2/с]
rA-гидрадинамический
радиус дифф. частицы; η-вязкость
растворителя; Клеточный эффект-явление,
состоящее в ограничении подвижности
частиц при дифф. перемещении, обусловл.
при налич. ближайшего окружения.
[см2/с]
rA-гидрадинамический
радиус дифф. частицы; η-вязкость
растворителя; Клеточный эффект-явление,
состоящее в ограничении подвижности
частиц при дифф. перемещении, обусловл.
при налич. ближайшего окружения.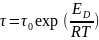 ,
,
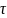 -время
пребыв. клетки,
-время
пребыв. клетки,
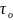 -период
колебания,
-период
колебания,
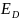 -энергия
активации диффузии.
-энергия
активации диффузии.D=D0exp(-ED/RT), η↑-больше затраты энергии требуется; ED
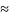 ΔHисп/3.
ΔHисп/3.Механизм взаимодействия частиц в «молекулярных клетках»
Классификация р-ий:1. диффузионно контролируемые/быстрые – процесс ограничен только коэф. диффузии D≈1/η; 2. псевдодиффузионные-необходима ориентация частиц ЕА≈0; 3. медленные- скорость не зависит от вязкости растворителя: ЕА>40 кДж/моль.
Диффузионный предел константы скорости р-ии
Ур-е для скорости р-ии: r=kCАCВ=4π(RA+RB)(DA+DB)CACB [молекул/м3с2] ;
kB=r/CACB [м3/моль*с]
kB=4π103*NARABDAB [л/моль*с] – для расчета диффузионного предела константы скорости р-ии. RAB=RA+RB, DAB=DA+DB
коэф. диффузии: D=
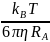 ур-е Стокса: DAB=
ур-е Стокса: DAB=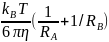
тогда ур-е можно упростить: kB=4π*103(RA+RB)*(kBT/6πη)*((1/RA)+(1/RB))NA=
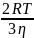 *
*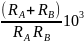
если RA=RB, DA=DB тогда: kB=(8RT/3η)*103
Температурная зависимость: η=η0exp(B/RT), В(Е-энергия активации вязкого течения)
Чаще всего: kэксп kD тк ЕА>0(не каждое столкновение приводит к образованию продукта р-ии)