

Заключение
В работе исследованы зависимости параметров электродного материала суперконденсатора от концентрации электропроводящего наполнителя (многослойных УНТ) и полимерного связующего. Несмотря на изоляционные свойства полимерного связующего с низкой диэлектрической проницаемостью, вклад в общее сопротивление не является столь значительным и его можно компенсировать увеличением содержания электропроводящего наполнителя. С ростом концентрации электропроводящего наполнителя у всех групп образцов наблюдается увеличение удельной емкости в растворе 0.1М H2SO4.
Литература
1. Carbon properties and their role in supercapacitors / A.G. Pandolfo, A.F. Hollenkamp // Journal of Power Sources 157 (2006) 11 - 27.
2. KOH activated carbon fabrics as supercapacitor material / K. Babel, K. Jurewicz // Journal of Physics and Chemistry of Solids 65 (2004) 275-280.
3. Capacitance limits of high surface area activated carbons for double layer capacitors / O. Barbieri, M. Hahn, A. Herzog, R. Kotz // Carbon 43 (2005) 1303-1310.
4. Conductivity percolation in carbon-carbon supercapacitor electrodes / N.L. Wu, S.Y. Wang // Journal of Power Sources 110 (2002) 233-236.
5. Небольсин В.А., Воробьев А.Ю. Роль поверхностной энергии при росте углеродных нанотрубок в процессе каталитического пиролиза ацетилена // Неорганические материалы. 2011. Т.47. №2. С. 168-172.
ОАО “Воронежское специальное конструкторское бюро «Рикон»”
УДК 621. 367. 502.7
И. М. Винокурова, С. А. Казакова
ИСПОЛЬЗОВАНИЕ АЛГОРИТМА ВНЕДРЕНИЯ ЭХО В ПРОИЗВОДСТВЕ ПРИ АНОДНОЙ ОБРАБОТКЕ
В материалах статьи приводятся данные по определению методов расчета процессов анодной обработки
Накопленный в авиастроении опыт применения ЭХО показывает, что для большинства технологических приложений пульсирующим режимом можно управлять, если установить закономерности его изменения. При значительном изменении по длине канала его ширины и плавности профиля задача расчета параметров течения рабочей среды сводится к двухмерной и даже одномерной схеме с переменной скоростью движения. Это наблюдается визуально при скоростной съемке [1-3]. Большое влияние на протекание процесса оказывает обоснованный учет фазового состава рабочей среды в зазоре (табл. 1).
Анализ табл. 1 и результатов скоростной съемки [2, 3] дает основания считать, что для абсолютного большинства приложений ЭХО в ракетно-космической технологии справедливо принять рабочую среду как 2 фазную, содержащую жидкую и газообразную составляющую и протекающую в пульсирующем режиме с частотой от 1 до сотен и тысяч циклов за время обработки. Из [4] видно, что гидроокислы в зазоре проявляются в виде загрязнений, достаточно равномерно заполняющих пространство. Это может быть объяснено возможностью нахождения такой фазы в жидкости в виде растворенных частиц, которые только после выхода из зазора переходят в нерастворимое состояние. Подобные исследования, выполненные с применением коагуляторов, показали образование в зазоре скоплений нерастворимых продуктов обработки, что, возможно, будет иметь место при формообразовании длинномерных поверхностей. Однако на технологические показатели гидроокислы оказывают незначительное влияние (по [4] не более 7 %), хотя могут повышать вязкость среды в зазоре.
Таблица 1
Влияние фазового состава рабочей среды на стабильность процесса ЭХО
Состав рабочей среды
|
Содержание в рабочей среде, %
|
Влияние на процесс ЭХО
|
Влияние на характер течения рабочей среды
|
|
|
начало канала
|
по длине канала
|
|
|
Жидкие рас- творы ней- тральных со- лей в воде |
100
|
снижа- ется
|
значи- тельное
|
незначи- тельное
|
Твердые частицы
|
возникают как результат загрязнения рабочей среды
|
влияют при концентрации более 5 %
|
||
Коллоидные гидроокислы |
отсутст- вуют |
до 8-10
|
незначи тельное |
незначи- тельное |
Газообразные продукты обработки
|
отсутствуют
|
до 100
|
значительное
|
значительное при длине канала до 200 зазоров опре-деляющее при длине канала свыше 200 зазоров |
На стадии отработки технологичности требуется обосновать вариант течения жидкости через рабочее пространство. Исходные данные здесь выбираются из карт заготовки и детали, что дает 3 варианта обработки: когда профиль заготовки близок к заданному в детали (рис. 1, а), в случае образования между электродами (профиль канала имитируют заготовка и катод-инструмент) диффузорного (рис. 1, б) или конфузорного (рис. 1, в) пространства.
 Рис.1.
К выбору направления подачи электролита
в зону обработки:
Рис.1.
К выбору направления подачи электролита
в зону обработки:
1 - заготовка (пунктиром показан профиль детали); 2 - электрод-инструмент; V - скорость прокачки электролита; So - начальный межэлектродный зазор
Равномерный зазор (рис. 1, а) в практике встречается редко, т.к. с позиций технолога в этом случае размерная предварительная обработка должна быть исключена, как не имеющая смысла, но вызывающая дополнительные затраты. Достаточно рассчитать припуск только на чистовую операцию, как правило, безразмерную. Поэтому будут анализироваться схемы "б, в" (рис. 1), где карты припусков формируют диффузорный и конфузорный каналы (при большой длине канала могут быть участки с обоими профилями). Для ЭХО, особенно на этапе удаления основной части припуска, целесообразно иметь схему "б" (рис. 1), поскольку она способствует лучшему массовыносу при выбранном направлении течения рабочей среды (по мере накопления продуктов обработки в зазоре сечение канала возрастает). Поэтому в варианте "в" (рис. 1) целесообразно изменить направление течения электролита (V) на противоположное и учесть это в техническом задании на технологическую оснастку. Если длина (по направлению течения рабочей среды) обрабатываемой поверхности не значительна (по [5] не свыше 200 межэлектродных зазоров So), то нижний предел скорости течения электролита находится из условия выноса продуктов обработки из зазора по осредненным (приближенным) зависимостям, например из [6].
Верхний предел зависит от геометрии канала. Если канал плоский или с аэродинамическим профилем, имеет незначительное плавное изменение ширины, то оборудование позволяет создать на входе однородный по давлению поток, а верхний предел скорости может быть выбран исходя из возможностей гидравлического агрегата (давление на входе, расход). Это позволяет снизить начальный межэлектродный зазор до предела, соответствующего началу образования пульсаций (с учетом возможностей системы регулирования зазора на оборудовании), повысить напряжение на электродах (не превышая предела, вызывающего пробой промежутка между электродами) и достичь наиболее высокой скорости обработки, точности, качества поверхности. Отсюда вытекает ранее не применяемая в технологии ЭХО научная концепция: для технологичных с позиции ЭХО деталей (по геометрии, припуску, длине канала, схеме подачи электролита) возможно путем оптимизации комплекса зазор - скорость электролита - напряжение на электродах достичь наилучших технологических показателей по производительности, точности, качеству поверхности. Если учесть, что по [7] только отклонения от рабочего режима течения электролита, влияющие также на стабильность его параметров, определяют более 20 % погрешностей обработки, а с учетом стабилизации на современных станках зазора - до 80 % погрешностей, то научно обоснованные режимы течения дают возможность получить для значительной группы деталей авиационной и космической техники наиболее высокие технологические результаты от применения ЭХО.
Использование разработанного алгоритма дает возможность рекомендовать внедрение ЭХО в производство для выполнения работ, перечисленных ниже:
1. Выполнение уникальных операций, осуществимых только электрохимическим методом. В этом случае нет необходимости обосновывать целесообразность применения ЭХО, так как иначе вообще невозможно изготовить разработанную деталь.
2. При изготовлении деталей из высокопрочных материалов практически нет необходимости учитывать их форму и масштаб выпуска. Здесь электрохимический метод обработки внутренних поверхностей имеет неоспоримые преимущества.
3. Для обработки заготовок из материалов, обладающих особыми свойствами (повышенной вязкостью, хрупкостью и др.), при обработке которых возникают технологические затруднения. К этой группе можно отнести жаропрочные, титановые и магнитные сплавы. Однако здесь имеются ограничения: если детали простой формы, а выпуск их ограничен, то внедрять процесс ЭХО, как правило, невыгодно. Например, простые втулки целесообразно изготовлять с применением ЭХО в случае, когда количество одноименных деталей обеспечивает загрузку хотя бы одного станка в течение смены без перенастройки.
4. Изготовление деталей из конструкционных сталей, которые наиболее широко используют в машиностроении. Такие операции рентабельны для длинномерных отверстий (l/(d> 810), при условии, если можно обеспечить загрузку станка с периодической перенастройкой не чаще 6-10 раз в течение месяца. Это соответствует масштабу выпуска 50-100 одноименных труб или 10-20 деталей с внутренней полостью переменного сечения. В случае, когда электрохимическое оборудование уже имеется на заводе и требуется только его переналадка, указанные цифры уменьшаются в 1,5-2 раза.
5. При обработке внутренних поверхностей в деталях из сплавов легких и цветных металлов (алюминия, меди, магния и др.) процесс ЭХО применим, если доступ инструмента к месту удаления припуска затруднен, а число изготовляемых деталей значительно (загрузка одноименными деталями не менее 1-3 смен). Создавать новое оборудование для таких деталей в большинстве случаев нецелесообразно, кроме деталей, требующих выполнения уникальных операций.
Для случая течения рабочей двухфазной среды с переменной скоростью в зазоре с различным профилем можно разработать физическую модель и сформулировать закон управления подачей среды в зависимости от геометрии конкретной обрабатываемой поверхности и способа регулирования межэлектродного зазора. По физической модели можно сформировать математическое описание процесса и реализовать его с использованием современных средств регулирования и управления (процессоры, блоки ЧПУ), имеющихся на оборудовании.
Литература
1. Электрохимическая размерная обработка. Проблемы и решения/ Г.Н. Зайдман // Электрохимическая размерная обработка. 1991. № 1. С. 3-14.
2. Саушкин Б. П. Электрохимическая обработка изделий из титановых сплавов / Б. П. Саушкин, Ю. Н. Петров, А. З. Нистрян, А. В. Маслов // Кишинёв. Штиница. 1988. 200 с.
3. Мандрыкина И. М. Исследование взаимосвязи термокине-тических и электрохимических параметров при импульсных режимах обработки титановых сплавов. Дис… Воронеж. 1998. 210 с.
4. Шалимов Ю. Н. Оптимизация электрохимического процесса обработки алюминиевой фольги в производстве конденсаторов / Ю. Н. Шалимов, И. М. Мандрыкина, Ю. В. Литвинов Воронеж: Изд-во ВГТУ, 2000. 343 с.
5. Винокурова И. М., Смоленцев В. П., Математическое описание процессов переноса в турбулентных потоках при электрохимиической обработке металлов. Современная электротехнология в промышленности центра России. Сб. тр. VIII региональ. научно-техн. конф. Тула, 1 июня 2006. –Тула:ТулГУ С. 22-26.
6. Смоленцев В. П. Технология электрохимической обработки внутренних поверхностей. Москва. Машиностроение. 1978. 176 с.
7. Газизулин К. М. Электрохимическая размерная обработка крупногабаритных деталей в пульсирующих рабочих средах: Научное издание. Воронеж: Воронежский государственный университет, 2002. 243 с.
Воронежский государственный технический университет
УДК 541.183
В.П. Горшунова, А.В. Рыльков, О.В. Чибисова
АДСОРБЦИЯ АММИАКА ТЕРМИЧЕСКИ МОДИФИЦИРОВАННЫМИ КРЕМНЕЗЕМНЫМИ СОРБЕНТАМИ
Приведены результаты исследования адсорбции аммиака термически активированными силикагелями разной пористости и аэросилом и дана сравнительная оценка поглотительных свойств
Аммиак, как известно, относится к аварийно химически опасным веществам. Аварии с выбросом NH3 происходят достаточно часто. Основная опасность аммиака в большей степени обусловлена возможностью его распространения и вредного воздействия на окружающую среду и людей. Поэтому проблемы улавливания аммиака в вентиляционных выбросах, а также защиты органов дыхания во время аварийных выбросов являются очень актуальными.
Нами изучено поглощение аммиака с использованием силикагелей марок КСКГ (крупнопористый) и КСМГ (мелкопористый) и аэросила. Выбор сорбентов был обусловлен тем, что по химической природе силикагель и аэросил относятся к кремнеземам, имеющим формулу SiO2, но разное строение.
Силикагели по своей химической природе представляют собой гидратированные аморфные кремнеземы (SiO2·nH2O), являющиеся реакционноспособными соединениями переменного состава, превращения которых происходят по механизму реакции поликонденсации:
n Si(OH)4 → SnnO2n-m + (2n-m) H2O.
Поликонденсация ведет к формированию структурной сетки сфера подобных частиц коллоидных размеров (от 2∙10-9 до 2∙10-8 м), сохраняющейся при высушивании гидрогеля кремниевой кислоты и образующей жесткий кремнекислородный каркас. Зазоры между частицами образуют пористую структуру силикагеля. Для получения силикагелей в промышленности обычно используют метод осаждения аморфного кремнезема из силикатов щелочных металлов минеральными кислотами.
Кремний содержащим сырьем является твердый силикат натрия, а в качестве реагента – минеральной кислоты – используется, как правило, наиболее дешевая серная кислота:
Na2O·3SiO2 + H2SO4 = 3SiO2 + H2O + Na2SO4.
Выпускают силикагели в виде шариков, таблеток или кусочков неправильной формы. Размеры их зерен составляют от 0,1 до 7,0 мм. Адсорбционные и химические свойства силикагелей существенно зависят от наличия на их поверхности групп ≡ Si – OH.
Силикагели служат для поглощения полярных веществ. Мелкопористые силикагели используют для адсорбции легкоконденсируемых паров и газов, крупнопористые и частично среднепористые силикагели служат эффективными поглотителями паров органических соединений. Высокое сродство поверхности силикагелей к парам воды обусловливает широкое их использование в качестве агентов осушки разнообразных газовых сред [1].
Аэросил представляет собой очень чистый аморфный непористый диоксид кремния SiO2 с размером частиц от 4 до 40 мкм. Это чрезвычайно легкий белый порошок. Снимки, сделанные при помощи электронного микроскопа, показывают, что аэросил состоит из частиц сферической или почти сферической формы, группирующихся в цепочки, которые в свою очередь, образуют хлопьевидные агрегаты. Рентгенографические исследования указывают на аморфную структуру. При истинном удельном весе 2,2 г/см3 насыпной вес порошка аэросила колеблется от 0,02 до 0, 05 г/см3.
По специальному заказу производится аэросил с высокой концентрацией диоксида кремния, в котором содержание примесей может быть доведено до любых минимальных пределов.
Особенность аэросила заключается в его большой удельной поверхности, обусловленной методом производства. Аэросил получают в результате гидролитического разложения тетрахлорида кремния SiCl4 в пламени водорода при температуре от 1100 до 1400 0С по реакции:
2H2 + O2 = 2H2O;
SiCl4 + 2H2O = SiO2+ 4HCl.
Для удаления хлористого водорода, адсорбированного поверхностью, аэросил обрабатывают влажным горячим воздухом.
Пористость у аэросила отсутствует. Экспериментально это доказывается тем, что удельная поверхность рассчитанная по газовой адсорбции, практически равна поверхности, которая определяется по электронно-оптическим снимкам. Величина частиц и характеристика готового продукта зависит от температуры пламени, состава газа и скорости потока на выходе из горелки. После прохождения пламенной зоны аэросил образуется сначала в виде аэрозоля обезвоженной кремниевой кислоты, который в дальнейшем проходит процесс коагуляции и образует звенья и хлопья сечением от 1 до 2 мкм.
Аэросил обладает хорошими адсорбционными свойствами. Вода, из-за своей способности образовывать с поверхностью аэросила водородные мостики, адсорбируется особенно хорошо. В 1 г аэросила с поверхностью 200 м2/г содержится примерно 1 моль групп ≡ SiOH, т.е. около 2000 групп ≡ SiOH на одну частицу аэросила [2].
Анализ
литературных данных позволяет
предположить, что и силикагель, и аэросил
можно использовать в качестве сорбентов
аммиака, так как аммиак является полярным
соединением, склонным к образованию
водородных связей с сорбентом. Величина
его дипольного момента (![]() =
1,46 D)
близка величине дипольного момента
воды (
=
1,46 D)
близка величине дипольного момента
воды (![]() .=
1,84 D).
Следовательно, возможна как адсорбция
аммиака, так и адсорбция воды.
.=
1,84 D).
Следовательно, возможна как адсорбция
аммиака, так и адсорбция воды.
Изучалась адсорбция в статических условиях, т.е. адсорбция проводилась из одного и того же объема газа вплоть до установления равновесия. Адсорбционная активность определялась количеством адсорбата, поглощенного единицей массы адсорбента при установлении равновесия гравиметрическим методом [3].
Для изучения процесса адсорбции создавались атмосферы с содержанием аммиака по объему, равными 20, 50, 100, 200 и 400 мг/м3. Для этого готовились растворы аммиака разной концентрации в соответствии с данными справочника [4]. В этих условиях концентрация водяных паров от 1,3 до 1,6 раза меньше, чем аммиака. Следовательно, можно с уверенностью предположить, что преимущественно сорбируется аммиак.
Сорбенты предварительно подвергали термической обработке. С целью подбора оптимальных условий температурного воздействия проводили следующий эксперимент: выдерживали сорбенты в сушильном шкафу при температурах 25, 50, 75, 100, 125, 150 и 175 0С и после каждой обработки изучали сорбционную емкость в газовой среде с объемной концентрацией аммиака 20 мг/м3 в течение 24 часов. Результаты приведены на рис.1, из которого следует, что наиболее подходящими условиями термообработки для сорбентов является температурный интервал от 110 до 120 0С.
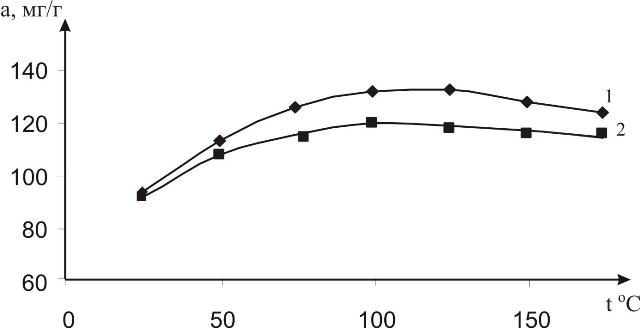
Рис. 1. Зависимость сорбционной активности сорбентов в газовой среде с объемной концентрацией аммиака, равной 20 мг/м3, от температуры обработки: 1 – силикагель марки КСКГ; 2 – аэросил
Далее изучали поглотительную способность термически обработанных от 110 до 120 0С сорбентов в вышеуказанных газовых средах и получили соответствующие изотермы адсорбции (рис.2).
Результаты опытов показали, что в исследованных условиях лучшими сорбционными свойствами обладает мелкопористый силикагель. Это связано с особенностями строения сорбентов. Известно [1], что на поверхности силикагелей имеются силанольные группы ≡ SiOH , которые при сближении друг с другом на расстояние 0,3 нм могут взаимодействовать друг с другом, образуя водородную связь. При этом образуется вицинальные группы. Эти поверхностные группы являются очень активными. Они могут образовывать водородную связь с полярными молекулами, в том числе с аммиаком. Так как удельная поверхность мелкопористого силикагеля значительно больше, чем крупнопористого (у КСМГ – 550 – 900 м2/г, а у КСКГ – 210 до 350 м2/г), то таких активных групп у мелкопористого силикагеля будет значительно больше, чем у крупнопористого. Отсутствие пор у аэросила в значительной степени снижает удельную поверхность (у аэросила удельная поверхность равна 200 м2 [2]), а следовательно, и концентрацию активных поверхностных групп.
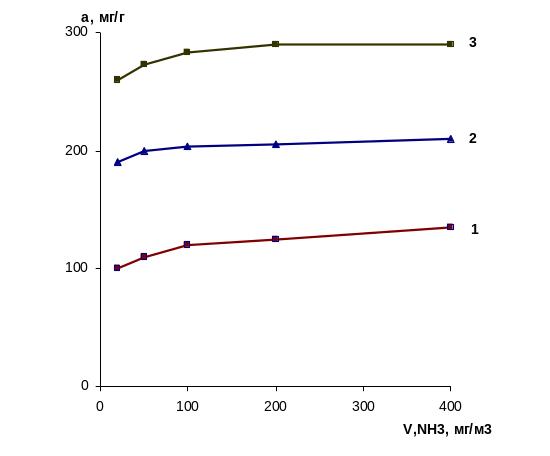
Рис. 2. Зависимость адсорбционной способности термически активированных сорбентов от объемной концентрации аммиака в газовой среде: 1 – аэросил; 2 - силикагель марки КСКГ; 3 – силикагель марки КСМГ
Таким образом, полученные результаты свидетельствуют о том, что в исследованных условиях лучшими поглотительными свойствами по отношению к аммиаку обладает термически модифицированный мелкопористый силикагель марки КСМГ.
Литература
1. Модифицированные кремнеземы в сорбции, катализе и хроматографии / Под ред. Г.В. Лисичкина. – М. : Химия, 1986. – 248 с.
2. Манченко Н.В. Аэросил, его свойства. применение и технические условия / Н.В. Манченко – Львов: Каменор, 1965. – 52 с.
3. Кельцев, Н.В. Основы адсорбционной техники. – М.: Химия, 1981. – 592 с.
4. Рабинович В.А. Краткий химический справочник / В.А. Рабинович, З.Я. Хавин. – С.- Пб.: Химия, 1997. – 392 с.
Воронежский государственный технический университет
УДК 621. 367: 502.7
И.М. Винокурова, А.А. Киселев
ОСОБЕННОСТИ ФОРМИРОВАНИЯ ОКСИДНЫХ ПЛЕНОК ПРИ АНОДНОМ ОКИСЛЕНИИ МЕТАЛЛОВ
Рассмотрены закономерности анодного растворения металлов при высоких плотностях с учетом особенностей пассивации
Образование оксида может происходить на поверхности металла, на поверхности или внутри оксидной пленки. Первый случай имеет место, если превалирует скорость диффузии кислорода (ионов или атомов), второй — если превалирует скорость диффузии ионов или атомов металла. В большинстве случаев скорости диффузии соизмеримы и зона роста оксидной пленки находится внутри, несколько ближе к ее внешней поверхности.
В соответствии с ионно-электронной теорией окисления, разработанной Вагнером, в оксидной пленке протекает встречная диффузия ионов металла и кислорода. При этом поверхность металла является анодной и на ней протекает реакция ионизации атомов металла. Образовавшиеся положительные ионы и освободившиеся электроны перемещаются в пленке раздельно (рис. 1). Электроны перемещаются с большей скоростью; диффузия же ионов металла протекает в результате перемещения либо по дефектным местам кристаллической решетки оксида, либо по ее междоузлиям.
Адсорбировавшиеся из газовой фазы молекулы кислорода диссоциируют на внешней поверхности оксида. Атомы кислорода, принимая электроны, движущиеся от поверхности металла, превращаются в ионы О2-, которые начинают диффундировать навстречу ионам металла. Таким образом, внешняя поверхность пленки, на которой кислород принимает электроны, является катодной поверхностью. Следовательно, встречная диффузия ионов металла и кислорода протекает в электрическом поле и кинетические уравнения, могут быть выведены, исходя из чисто электрических параметров и закономерностей: величин ионной и электронной проводимости, чисел переноса ионов и электронов, закона Ома.
В силу того, что радиусы ионов металла значительно меньше радиуса иона кислорода, скорость диффузии первых несколько выше. Это и является основной причиной того, что образование оксида (рост пленки) происходит в зоне, более близкой к внешней поверхности пленки.
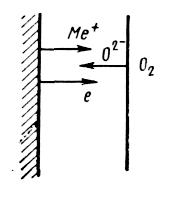 Рис.
1. Схема ионно-электронного механизма
Рис.
1. Схема ионно-электронного механизма
высокотемпературного окисления
В пассивном состоянии рост анодной плёнки на титане происходит по линейному закону с "константой анодирования" 2,6 нм/В.
При потенциале анода Eа = 2,7 В по отношению к нормальному каломельному электроду (н. к. э.) на аноде одновременно с окислением титана происходит и процесс окисления воды, то есть протекают одновременно два процесса
Ti + 2H2O - 4e TiO2 + 4H+, (1)
2H2O - 4e O2 + 4H+. (2)
Большая часть потенциала приходится на его падение в оксидной плёнке, лишь его незначительная часть локализуется в двойном электрическом слое [1]. По мнению исследователей [1], в пассивной области перед анодно-анионной активацией рост анодно-оксидной плёнки (аоп) осуществляется в два этапа - по мере увеличения Eа. В области значений стационарного потенциала до величины порядка 10 В происходит равномерный рост оксидной плёнки с одновременным "залечиванием" дефектов в ней. Дальнейшее увеличение потенциала приводит к снижению защитных свойств аоп, несмотря на возрастание её толщины, т.к. появляется возможность возникновения и развития пор, трещин. Формирующаяся оксидная плёнка в этом случае имеет аморфный характер. Присутствие в растворе электролита активирующих анионов Br-, J-, Cl-, ClO4- на этих этапах создаёт конкуренцию между процессом "залечивания" дефектов и процессом адсорбции активирующих анионов в местах дефектов, где происходит образование химической связи с поверхностными атомами металла и сольватация образовавшихся соединений с переходом их в раствор. При достижении потенциала питтингообразования (E п ) суммарный ток увеличивается во времени за счёт увеличения площади активированной поверхности электрода. Развитие активационного процесса идёт с малыми скоростями, но после активации значительной площади электрода наблюдается всплеск тока, характеризующий увеличение скорости процесса растворения металла [2]. Рост тока прекращается после охвата всей поверхности электрода активационным процессом. В потенциостатическом режиме, при развитии активационного процесса аоп, обладающая высоким электросопротивлением, удаляется с поверхности электрода. В режиме поддержания постоянного анодного потенциала резко возрастает анодный ток (иногда на несколько порядков), и в результате этого происходит значительный нагрев
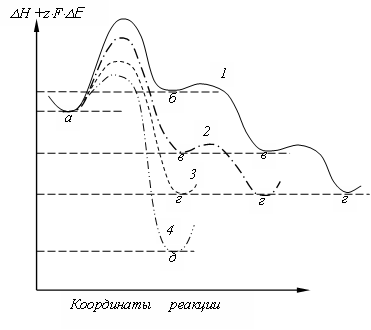
Рис.2. Схема изменения энергии в ходе анодного растворения металла (справа налево) с учетом электрической (кулоновской) части энергии: а, б, в, г, д – положении ионов металлов соответственно в электролите; ад-атом; в ступени роста; в полукристаллическом положении (месте роста); в верхнем заполненном атомном слое; 1, 2, 3, 4 – пути реакции [4]
поверхности электрода до TS= Ткр (критической температуры, при которой процесс становится неустойчивым).
Значения энергии различных состояний в ходе растворения металла показаны на рисунке 2. Состояние, а соответствует нахождению атома (иона) вне твердой фазы (металлической фазы), в положении б атом (ион) находится на плоскости решетки в виде ад - атома (ад - иона), в положении в - на кристаллической ступени и г – в полукристаллическом положении, которое называется также местом роста растворения кристалла. Положение д – соответствует верхнему заполненному атомному слою.
Растворение может, осуществляется разными путями, которые отличаются скоростью, а предпочтение в процессах перехода зависит от энергий активации отдельных стадий и от частоты осуществления этих состояний.
Число поверхностных позиций, которые имеют для анодной реакции перехода 1 (а б), всегда намного больше, чем для реакции перехода 2 (а в), и еще больше, чем число полукристаллических положений для реакции перехода 3 (а г). Так как необходимо принять, что энергии активации Ед Ев Ег Ед, как вытекает из рис. 2, то влияние меньшего числа поверхностных позиций на плотность тока обмена через снижение энергии активации более или менее компенсируется. Поэтому предположение о сравнимости величин плотностей тока обмена i0, 1, io, 2, io, 3 необходимо четко определять и соответственно учитывать пути реакции при рассмотрении экспериментальных данных. Для каждого процесса перехода ток обмена имеет свое конкретное значение, а именно: i0, 1 - ток обмена с химическим перенапряжением, io, 2 – ток обмена реакции с поверхностным перенапряжением, io, 3 - ток обмена реакции кристаллизации.
Первый энергетический пик, приведенный на диаграмме Н +zFЕ – координата реакции соответствует энергии активации электрохимической реакции.
Для наиболее вероятного случая растворения кристаллов через винтовые дислокации с параллельными ступенями растворения при оценке порядка величины омического падения напряжения в электролите нужно исходить из распределения линий тока вокруг линий растворения, как осей с цилиндрической симметрией.
При растворении вокруг винтовой дислокации как центра образуется ступень роста спиральной формы. В стационарном состоянии эта спираль вращается с постоянной угловой скоростью. Если принять, что радиальная скорость роста ступени не зависит
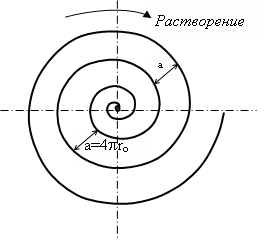
Рис. 3. Спираль роста по уравнению (3) r=2ro (спираль Архимеда) при осаждении (центр приподнят) или растворении (центр углублен) металла на одной винтовой дислокации
от расстояния r от центра спирали и от направления роста , то этому условию удовлетворяет спираль Архимеда, которая в полярных координатах описывается уравнением
r=2ro. (3)
Дифференцированием этого уравнения по t получаем
![]() (4)
(4)
а
отсюда не зависящую от r
и
угловую скорость вращения спирали
![]() (5)
(5)
Резюмируя результаты химического и электрохимического поведения оксидных плёнок на титане, определяющих скорость анодного процесса, можно сделать следующие выводы:
1. Электролиты для анодной обработки титана не должны содержать компонентов, инициирующих образование "барьерных слоёв" значительной толщины [5].
2. Концентрация окислителей в составе электролита должна соответствовать оптимуму, исключающему химический механизм растворения оксида титана по всей поверхности.
3. Рассеивающая способность электролита, определяющая равномерность распределения тока по поверхности обрабатываемого металла, выбирается из условий максимально допустимого отклонения по удельной ёмкости.
4. Химический состав электролита обеспечивает минимум электрических потерь, так как процесс электролиза протекает при достаточно высоких плотностях тока.
Литература
1. Давыдов А. Д., Козак Е. Высокоскоростное электрохимическое формообразование. М.: Наука. 1990. 272 с.
2. Румянцев Е. М., Давыдов А. Д. Технология электрохимической обработки материалов. М.: Высшая школа. 1984. 159 с.
3. Мандрыкина И. М. Исследование взаимосвязи термокине-тических и электрохимических параметров при импульсных режимах обработки титановых сплавов. Дис… Воронеж. 1998. 210 с.
4. Феттер К. Электрохимическая кинетика. Изд-во химия, 1967. 856 с.
5. Шалимов Ю. Н. Оптимизация электрохимического процесса обработки алюминиевой фольги в производстве конденсаторов / Ю. Н. Шалимов, И. М. Мандрыкина, Ю. В. Литвинов Воронеж: Изд-во ВГТУ, 2000. 343 с.
Воронежский государственный технический университет
УДК 541.183
В.П. Горшунова, А.В. Рыльков, О.В. Чибисова, В.А. Небольсин
АКТИВИРОВАННЫЙ УГОЛЬ В КАЧЕСТВЕ СОРБЕНТА АММИАКА
Представлены результаты исследования адсорбции аммиака медицинским углем, а также термически и термохимически модифицированным активным углем марки АГ-3. Показано, что наибольшей поглотительной способностью по отношению к аммиаку обладает термохимически модифицированный уголь АГ-3
Аммиак, является исходным сырьем для производства азотной кислоты, минеральных удобрений. Кроме того, он остается на сегодняшний день основным хладагентом в аммиачных холодильных установках (АХУ). Аммиак обладает токсическими свойствами. Он вызывает острое раздражение слизистых оболочек, слезоточение, ожоги, удушье. Предельно допустимая концентрация аммиака составляет: в воздухе рабочей зоны 20 мг/м3, в воздухе населенных пунктов максимально разовая и среднесуточная ПДК составляет 0,2 мг/м3 [1].
Технологические процессы, связанные с наличием промышленных токсических выбросов, предусматривают меры по их очистке. В связи с тем, что концентрация токсичных веществ в промышленных сбросах мала, наиболее рациональным (а иногда и единственным) методом очистки служит адсорбционный, с которым ни один из известных методов тонкой очистки большинства жидких и газообразных веществ не может конкурировать ни по стоимости, ни по эффективности [2].
Иногда примеси токсичных веществ, например, аммиака, появляются в воздухе рабочей зоны промышленных предприятий, на которых аммиак используется в качестве хладагента в аммиачных холодильных установках (АХУ), в результате разгерметизации отдельных систем и блоков установки. Наличие стационарных вытяжных вентиляционных систем на некоторых предприятиях приводит к очистке помещений рабочей зоны. Однако эти выбросы необходимо улавливать, чтобы они не попадали в жилую зону. Это можно осуществить с помощью подходящих адсорбентов, находящихся в специальных контейнерах и помещенных в систему газоотвода.
Задача изучения поглотительной способности наиболее доступного сорбента - активного угля - по отношению к аммиаку.
По своим структурным характеристикам активные угли относятся к группе микрокристаллических разновидностей углерода – это графитовые кристаллиты, состоящие из плоскостей протяженностью от 2 до 3 нм, которые в свою очередь образованы гексагональными кольцами. Однако типичная для графита ориентация отдельных плоскостей решетки относительно друг друга в активных углях нарушена. Слои беспорядочно сдвинуты и не совпадают в направлении, перпендикулярном их плоскости. Неоднородная масса, состоящая из кристаллитов графита и аморфного углерода, определяет своеобразную пористую структуру активных углей, а также их адсорбционные и физико-механические свойства.
Пористая структура активных углей характеризуется наличием развитой системы пор, которые классифицируют по размерам на микропоры, мезопоры и макропоры в табл. 1.
Таблица 1
Характеристика пор активных углей
Поры |
Размер, нм |
Микропоры |
Менее 1,6 |
Мезопоры |
1,6-200 |
Макропоры |
Более 200 |
Микропоры – наиболее мелкая разновидность пор, соизмеримая с размерами адсорбируемых молекул. Удельная поверхность микропор – от 800 до 1000 м2/г.
Мезопоры – поры, для которых характерно послойное заполнение поверхности, завершающееся их объемным заполнением по механизму капиллярной конденсации. Удельная поверхность мезопор может достигать от 100 до 200 м2/г.
Макропоры – самая крупная разновидность пор, удельная поверхность которых обычно не превышает от 0,5 до 0,2 м2/г. Макропоры в процессе адсорбции не заполняются, а выполняют роль транспортных каналов для доставки адсорбата к поверхности адсорбирующих пор.
Предварительная подготовка сырья - это приведение исходного угольного сырья в состояние, удобное для осуществления дальнейшей термической обработки. Карбонизация – это термическая обработка материала без доступа воздуха для удаления летучих веществ. На стадии карбонизации формируется каркас будущего активного угля – первичная пористость, прочность и так далее.
Активация водяным паром представляет собой окисление карбонизованных продуктов до газообразных в соответствии с реакцией С + Н2О = СО + Н2 или при избытке водяного пара С + 2 Н2О = СО2 + 2 Н2. В процессе активации развивается необходимая пористость и удельная поверхность. Происходит значительное уменьшение массы твердого вещества, именуемое обгаром.
В зависимости от назначения угли подразделяют на газовые, рекуперационные, осветляющие и угли-носители катализаторов-хемосорбентов.
В табл. 2 приведены характеристика и области применения некоторых марок активных углей.
Марка углей позволяет судить об их происхождении или назначении: АУ – активный уголь; БАУ – березовый активный уголь; АГ – гранулированный активный уголь; АР - активный уголь рекуперационный; СКТ – сернистокалиевого активирования, торфяной; КАУ – косточковый активный уголь; ОУ – осветляющий активный уголь.
Активные угли характеризуются гидрофобностью (плохой сорбируемостью полярных веществ, к которым принадлежит и вода). Это свойство определяет широкое их использование в практике рекуперационной и санитарной очистки отходящих газов разнообразной влажности.
По размеру и форме частиц активные угли подразделяют на гранулированные и порошкообразные. Гранулированные угли изготавливаются обычно в форме цилиндриков диаметром от 2 до 5 мм, причем высота цилиндрика всегда больше диаметра [2].
Таблица 2
Характеристика и области применения некоторых марок активных углей
Марка |
Размер гранул, нм |
Насыпная плотность, кг/м3 |
Предельный адсорбционный объем микропор, см3/г |
Области применения |
БАУ |
1 – 5 |
350 |
0,26 |
Адсорбция газов и паров |
СКТ |
1,5-2,7 |
380-450 |
0,45-0,56 |
- « - « - « - « - « - |
АГ-3 |
1,5-2,7 |
450 |
0,3 |
- « - « - « - « - « - |
АГ-5 |
1,0-1,5 |
450 |
0,3 |
- « - « - « - « - « - |
САУ |
1,0-5,0 |
450 |
0,36 |
- « - « - « - « - « - |
КАУ |
1,0-5,0 |
400 |
0,33 |
- « - « - « - « - « - |
АР-3 |
2,0-5,0 |
до 600 |
0,39 |
Для целей рекуперации |
КАД |
1,0-1,5 |
400 |
0,3 |
Для извлечения иода из растворов |
Медицинский уголь - это поглощающее средство, которое изготавливают из древесного и ископаемого угля. Специальный «лечебный» уголь изготавливают при помощи тепловой обработки исходного материала в вакуумных условиях.
Препарат получают из древесины методом сухой перегонки. Для этой цели используются деревья различных пород: бук, сосна, липа, дуб, ель, осина, ольха, тополь. Сегодня активированный уголь применяется в медицине в основном как универсальный антидот для поглощения различных ядов – бактериального, грибкового, растительного, животного или химического происхождения.
В данной работе использовали активный уголь марки АГ-3 и медицинский уголь. Указанные сорбенты применяли с целью поглощения аммиака.
Исследование проводили в условиях, максимально приближенным к реальным условиям, возникающим при выбросах аммиака и нейтрализации его с помощью водяных завес: готовились растворы аммиака разной концентрации в соответствии с данными справочника [3]. Объемные концентрации аммиака рассчитывали по уравнению Клапейрона-Менделеева.
Адсорбция изучалась в статических условиях из одного и того же объема газа вплоть до установления равновесия. Адсорбционная емкость сорбентов определялась количеством адсорбата, поглощенного единицей массы адсорбента при установлении равновесия. Использовался классический метод гравиметрии при Т = 298 К [4].
Уголь АГ-3 предварительно активировали «острым паром» в течение часа. Медицинский уголь измельчали с целью увеличения площади соприкосновения с адсорбатом.
Для изучения процесса адсорбции создавались атмосферы с содержанием аммиака по объему, равным 20, 50, 100, 200, 400 мг/м3, в которых сорбенты выдерживались до установления равновесия. Получали кинетические зависимости в координатах а, мг/г адсорбента – t, час. Подобная зависимость приведена на рис. 1 для объемной концентрации 100 мг/м3.
На основе полученных данных можно сделать вывод: в изученных условиях большая адсорбционная способность наблюдалась у активного угля марки АГ-3 по сравнению с медицинским углем. Эти результаты, полученные на адсорбентах, поступающих в продажу, позволяют предположить, что их сорбционные свойства можно улучшить путем модифицирования.
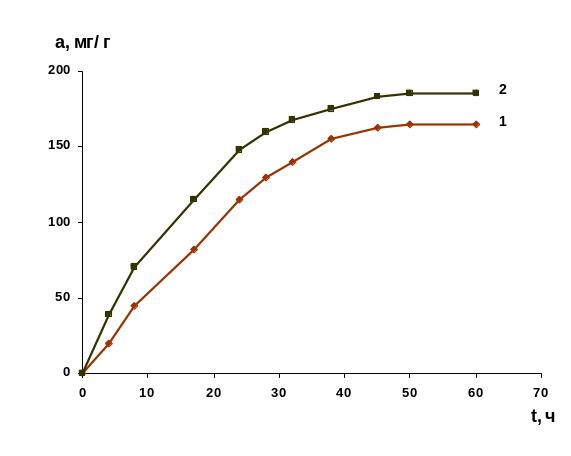
Рис. 1. Зависимость адсорбции аммиака от времени при его объемной концентрации в атмосфере 100 мг/м3: 1 – медицинский уголь; 2 – активный уголь АГ-3
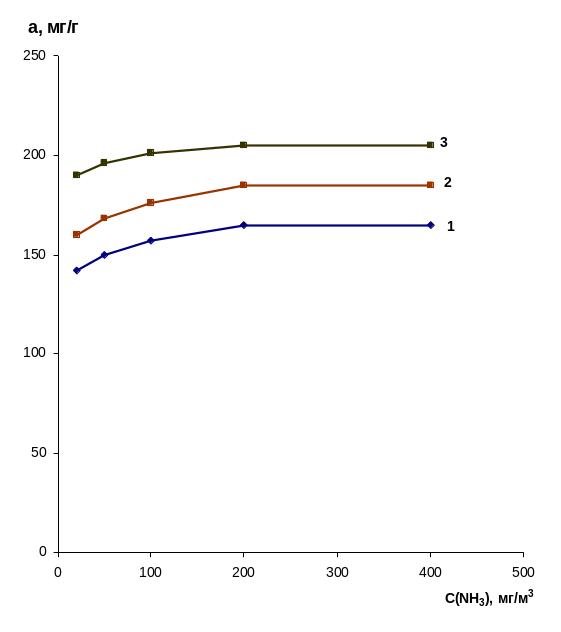
Рис. 2. Зависимость адсорбции углей разного типа от объемной концентрации аммиака: 1 – медицинский уголь; 2 – активный уголь АГ-3;
3 – термохимически модифицированный уголь АГ-3
Следующая серия опытов была проведена с активным углем АГ-3, предварительно химически активированного раствором сульфата меди. Насыщение осуществляли в 0,1 М растворе CuSO4 в течение 24 часов при постоянном перемешивании. Поглотительные свойства химически модифицированного активного угля изучали в атмосфере аммиака с объемными концентрациями 20, 50, 100, 200 и 400 мг/м3. Полученная изотерма адсорбции приведена на рис.2.
На основании данных результатов можно заключить, что химическое модифицирование активного угля марки АГ-3 повышает его поглотительную способность. Если сравнивать адсорбционные свойства термохимически активированных углей с силикагелями [5], то оказывается, что силикагели обладают большими поглотительными свойствами. Это объясняется разным механизмом сорбции. В угле АГ-3 преобладают мелкие и средние поры, в которых действуют физические силы между молекулами аммиака и сорбентом. В химически модифицированном угле к этим взаимодействиям добавляется донорно-акцепторное связь с катионами меди. В силикагелях, особенно в мелкопористом марки КСМГ, помимо вышеназванных взаимодействий в большей мере сказывается образование водородных связей между молекулами NH3 и активными структурными группами (силанольными и вицинальными) на их поверхности.
Литература
1. Лазарев Н.В. Вредные вещества: справочник / Н.В. Лазарев и др.; под ред. Н.В. Лазарева. – М.: Химия, 1971. – 142 с.
2. Лукин В.Д. Адсорбционные процессы в химической промышленности / В.Д. Лукин. – Л.: Химия, 1983. – 64 с.
3. Рабинович В.А. Краткий химический справочник / В.А. Рабинович, З.Я. Хавин. – С.- Пб.: Химия, 1997. – 392 с.
4. Кельцев, Н.В. Основы адсорбционной техники. – М.: Химия, 1981. – 592 с.
5. В.П. Горшунова, С.С. Шмакова, В.А. Небольсин. Адсорбция аммиака термохимически модифицированными силикагелями разной пористости.- Вестник ВГТУ, 2011, т.7, № 12, с. 51-53.
Воронежский государственный технический университет
УДК 502
И.Е. Рохас Риоха
ОСНОВНЫЕ ПОСЛЕДСТВИЯ ЗАГРЯЗНЕНИЯ АТМОСФЕРЫ
НЕКОТОРЫМИ СОЕДИНЕНИЯМИ АЗОТА
Обсуждаются последствия загрязнения атмосферы соединениями азота. Описываются методики определения концентрации оксидов азота и аммиака спектрофотометрическим методом. Анализируются способы очистки отходящих газов от некоторых соединений азота
Химическое загрязнение воздуха представляет огромную опасность для человечества. Вещества, попадающие в атмосферу в результате хозяйственной деятельности, не просто изменяют ее состав, но оказывают существенное влияние на сложившиеся в результате длительного развития процессы, протекающие в ней. Значительное негативное воздействие на состояние атмосферного воздуха оказывают газообразные и аэрозольные выбросы промышленного и бытового происхождения. По сравнению с другими компонентами окружающей среды, атмосфера характеризуется большей пространственной мобильностью и загрязняется наиболее быстро. Результаты экологических исследований свидетельствуют о том, что загрязнение воздуха – самый мощный и постоянно действующий фактор, оказывающий влияние на здоровье человека и других живых организмов. Значительное место среди загрязнителей атмосферы занимают различные соединения азота. Почти все летучие соединения азота относятся к ядовитым. Например, аммиак NH3 раздражает верхние дыхательные пути и органы зрения, а в высоких концентрациях оказывает возбуждающее действие на центральную нервную систему. Веселящий газ N2O в больших концентрациях в смеси с кислородом действует как наркотик. Оксид азота NO ведет к превращению гемоглобина крови в метгемоглобин и влияет на центральную нервную систему [5]. Диоксид азота NO2 приводит к отеку легких [2]. Эфиры азотистой кислоты расширяют сосуды, снижают давление и переводят гемоглобин крови в метгемоглобин. Эфиры азотной кислоты обладают наркотическими свойствами, поражают сердечно-сосудистую систему, а также приводят к образованию метгемоглобина.
Аммиак NH3 применяется в производстве азотной кислоты, нитрата и сульфата аммония, жидких удобрений (аммиакатов), мочевины, соды, в органическом синтезе, при крашении тканей, при серебрении зеркал. Высокие концентрации этого газа вызывают у человека слезотечение и боль в глазах, приступы кашля, головокружение, боль в желудке. К последствиям острого отравления аммиаком относятся помутнение хрусталика, потеря голоса, хронический бронхит. Порог восприятия этого газа составляет 0,037 мг/л. Предельно допустимая концентрация аммиака равна 20 мг/м3.
Для измерения концентрации аммиаки в технологических газах в диапазоне 0,2 – 5 мг/м3 широко применяется методика с реактивом Несслера. Метод измерения основан на образовании окрашенного в желтый цвет вещества при взаимодействии аммиака с реактивом Несслера и последующем определении содержания аммиака спектрофотометрическим методом. Измерению мешают аммонийные соли, некоторые амины алифатического ряда, формальдегид концентрацией больше 0,2 мг/м3, сероводород концентрацией больше 0,4 мг/м3, хлор концентрацией больше 0,2 мг/м3. Оптическую плотность раствора определяют при длине волны =450 нм.
Фторид азота NF3 применяется в качестве фторирующего агента и сырья для получения тетрафторгидразина и других фторидов азота. Вдыхание этого газа при его повышенных концентрациях вызывает у человека головную боль. Предельно допустимая концентрация фторида азота составляет 29 мг/м3.
Очень опасными загрязнителями атмосферного воздуха являются оксиды азота. Эти вещества составляют приблизительно 1/3 часть всех токсичных выбросов промышленного происхождения. В природе оксиды азота могут попадать в атмосферу во время лесных пожаров. Высокие концентрации оксидов азота в городах и окрестностях промышленных предприятий связаны с хозяйственной деятельностью человека [6]. В большом количестве оксиды азота поступают в атмосферу в результате работы теплоэлектростанций и двигателей внутреннего сгорания [7]. Оксиды азота образуются при синтезе азотной кислоты, во время процессов, связанных с получением мышьяковой кислоты и арсената натрия, серной кислоты по нитрозному способу, с получением щавелевой и хромовой кислот, алифатических и ароматических нитросоединений, анилиновых красителей; при изготовлении фотопленки, искусственного шелка, при травлении металлов; при взрывных работах в угольных шахтах, при сварке, плазменной резке металлов; при силосовании зерна. Влияние воздуха, загрязненного оксидами азота, на здоровье человека можно разделить на два основных вида в зависимости от времени проявления эффекта: 1) острое, появляющееся в период или непосредственно после повышения концентрации токсического вещества; 2) хроническое, результат которого сказывается не сразу, а через некоторое время. В обоих этих случаях загрязнение атмосферы может являться как причиной заболеваний, так и оказывать неспецифическое отягощающее воздействие, т. е. выступать в качестве провоцирующего фактора [3]. При контакте оксидов азота с влажной поверхностью легких образуются азотная HNO3 и азотистая HNO2 кислоты, поражающие альвеолярную ткань, что приводит к отеку легких [1]. При отравлении оксидами азота в крови человека возникают нитраты и нитриты. Нитриты вызывают расширение кровеносных сосудов и снижение давления. Они ведут к превращению оксигемоглобина в метгемоглобин, что нарушает снабжение кислородом клеток организма. При острых отравлениях большими концентрациями оксидов азота возможен смертельный исход. В случае хронических отравлений ими у человека возникают хронические воспалительные заболевания верхних дыхательных путей, хронические бронхиты, разрушаются коронки зубов. Иногда у людей, подвергшихся воздействию высоких концентраций оксидов азота, может появляться желтоватое окрашивание на коже кистей рук и ноздрей.
Веселящий газ N2O используется в качестве исходного продукта для получения азида натрия; в медицине применяется в смеси с кислородом для анестезии при хирургических операциях. В высоких концентрациях этот газ вызывает удушье вследствие вытеснения кислорода из легких. Газовые смеси, содержащие 20 %, 30 % и 50 % веселящего газа ухудшают кратковременную память у человека.
Оксид азота NO образуется в качестве промежуточного продукта при получении азотной кислоты окислением аммиака или азота воздуха. На начальной стадии острого отравления этим газом появляется общая слабость, головокружение, онемение ног. Затем снижается кровяное давление. При тяжелой форме отравления им замедляется пульс, изменяется цвет крови по причине образования метгемоглобина из оксигемоглобина, увеличиваются печень и селезенка. Оксид азота способен вызывать паралич и судороги. Последствия отравления им выражаются в ослаблении памяти и мышечной силы.
Диоксид азота NO2 образуется при использовании концентрированной азотной кислоты, например, при нитровании глицерина, при травлении меди и медных сплавов. Этот газ применяется в качестве гомогенного катализатора для получения серной кислоты камерным способом. Диоксид азота оказывает сильные раздражающее действие на дыхательные пути, особенно глубокие, что приводит к развитию токсического отека легких [4, 5]. Он угнетает аэробное дыхание и стимулирует анаэробное окисление в легочной ткани. Предельно допустимая концентрация для диоксида азота равна 9 мг/м3, для оксида азота – 30 мг/м3.
Широкое применение для измерения концентрации оксидов азота нашел спектрофотометрический методом с использованием реактива Грисса – Илосвая, предназначенный для определения содержания суммы оксида азота и диоксида азота в диапазоне концентраций 10 – 1000 мг/м3. В его основе лежит взаимодействие нитрит-иона и n-аминобензолсульфокислоты (сульфаниловой кислоты) с образованием диазосоединения, которое, реагируя с 1-нафтиламином, дает азокраситель, окрашивающий раствор от бледно-розового до красно-фиолетового цвета. Интенсивность этой окраски пропорциональна содержанию нитритов. Измерению мешает диоксид серы. Для устранения его влияния при отборе газа перед газовой пипеткой помещают трубку с кристаллическим оксидом хрома (VI) и добавляют в поглотительный раствор 10 % ацетона. Оптическую плотность раствора измеряют при длине волны =540 нм.
Оксиды азота участвуют в образовании фотохимического смога. Этот тип смога возникает летом при интенсивном воздействии солнечного света на воздух, чрезмерно насыщенный выхлопными газами автомобилей. Для появления фотохимического смога в атмосфере должны присутствовать оксиды азота и углеводороды. При слабом движении воздуха или безветрии в воздухе в солнечную погоду протекают сложные реакции с образованием новых токсичных загрязнителей – фотооксидантов, например, пероксиацетилнитратов, которые раздражают слизистые оболочки желудочно-кишечного тракта, дыхательной системы, органов зрения, а также могут вызвать гибель растений.
Промышленные выбросы оксидов азота являются одной из причин выпадения кислотных дождей. Оксиды азота соединяются с атмосферной влагой, и образуется разбавленная азотная кислота:
2NO + O2 2NO2,
NO2 + OH HNO3.
Под влиянием кислотных осадков из почвы выщелачиватются токсичные тяжелые и легкие металлы: свинец, кадмий, алюминий. Затем они или их токсичные соединения попадают в растения, что ведет к негативным последствиям.
Кроме того, оксиды азота в некоторой степени способствуют разрушению озонового слоя, попадая в верхние слои атмосферы при запусках космических кораблей и полетах сверхзвуковых самолетов. Считается, что одна молекула различных оксидов азота может разрушить около десяти молекул озона.
В настоящее время активно ведется разработка мероприятий, направленных на сокращение поступления газообразных соединений азота в атмосферу. Для предотвращения отравления оксидами азота на предприятиях можно применять фильтрующий промышленный противогаз, резиновые кислотостойкие перчатки, спецодежду, покрытую слоем перхлорвиниловой смолы. Проводится герметизация производственных процессов, в результате которых выделяются оксиды азота, осуществляется удаление образующихся оксидов азота местными вытяжными устройствами. При электро- и газовой сварке внутри аппаратуры требуется подача свежего воздуха для вытеснения оксидов азота, которые могут образовываться при окислении азота воздуха под воздействием высокой температуры. При плазменной резке металлов необходимо проведение автоматизации и механизации управления процессом, установка местных вентиляционных отсосов. При травлении металлов нужно установить местные отсосы от травильных ванн, добавлять вещества, поглощающие оксиды азота, создавать непроницаемый слой. На предприятиях необходимо вводить дистанционное управление технологическими процессами, связанными с образованием оксидов азота. Нужно устанавливать специальные фонтанчики и краны гидрантов для промывания глаз и кожи.
На предприятиях химического производства очистка промышленных газов от оксидов азота происходит в основном в результате превращений на катализаторах. Окислительные методы очистки воздуха основаны на реакции окисления оксидов азота с последующим поглощением водой и образованием азотной кислоты. Окисление озоном в жидкой фазе осуществляется по реакции:
2NO + O3 + H2O 2NHO3.
Окисление кислородом при высокой температуре происходит по реакции:
2NO + O2 2NO2.
Восстановительные каталитические методы очистки воздуха основаны на восстановлении оксидов азота до нейтральных продуктов в присутствии катализаторов или под действием высоких температур в присутствии восстановителей. Процесс восстановления отражает следующая схема:
![]() .
.
Разложение оксидов азота до нейтральных соединений (2NO N2 + O2) происходит в потоке низкотемпературной плазмы (10000 С). Этот процесс при более низких температурах в присутствии катализаторов осуществляется в двигателях внутреннего сгорания. Присутствие восстановителей в зоне реакции (угля, графита) тоже снижает температуру реакции восстановления.
При температуре выхлопных газов автомобиля в двигателях внутреннего сгорания возможно протекание реакции:
2NO + 2CO N2 + 2CO2.
Среди сорбционных методов очистки широкое применений имеют поглощение оксидов азота водными растворами щелочей и известью CaCO3 и адсорбция оксидов азота твердыми сорбентами, например, углем, торфом, силикагелем.
Литература
1. Голицын А.Н. Промышленная экология и мониторинг загрязнения природной среды / А.Н. Голицын. – М.: Оникс, 2007. 336 с.
2. Гусакова Н.В. Химия окружающей среды / Н.В. Гусакова. – Ростов н/Д: Феникс, 2004. 192 с.
3. Куликова Е.С. Загрязнение атмосферы оксидами азота / Е.С. Куликова // Экология Центрально-Черноземной области Российской Федерации. 2002. №2. С 116 – 118.
4. Новиков Ю.В. Экология, окружающая среда и человек / Ю.В. Новиков – М.: ФАИР-ПРЕСС, 2005. 736 с.
5. Общая токсикология / под ред. Б.А. Курляндского, В.А. Филова. – М.: Медицина, 2002. 608 с.
6. Трифонов К.И. Физико-химические процессы в техносфере / К.И. Трифонов, В.А. Девисилов. – М.: Форум, 2007. 240 с.
7. Экология и безопасность жизнедеятельности / под ред. Л.А. Муравья. – М.: ЮНИТИ, 2003. 350 с.
Воронежский государственный технический университет
УДК 621.438.2
М.В. Горковенко, И.С. Правдин, Л.М. Трофимов
НИКЕЛЕВЫЕ СПЛАВЫ ДЛЯ ПЕРСПЕКТИВНЫХ ГТД
В статье дан анализ перспективных жаропрочных сплавов на никелевой основе для лопаток газовых турбин авиационных ГТД
В настоящее время в России и за рубежом ведутся активные работы по созданию, освоению и доводке авиационных газотурбинных двигателей (ГТД) нового поколения, включая двигатели военного, пассажирского и транспортного назначения. Одним из главных требований к перспективным ГТД является повышение на 20-25 % топливной эффективности, что в свою очередь связано с необходимостью увеличения температуры газа на входе в турбину на 180-220 °С [1], а это возможно обеспечить используя в конструкции газовых турбин новые никелевые сплавы.
С начала 90-х годов за рубежом проводятся достаточно интенсивные исследования и разработки по созданию никелевых суперсплавов четвертого поколения, легированных рением и элементом платиновой группы - рутением. Проведенными к настоящему времени исследованиями установлено, что эти элементы повышают сопротивление сплавов высокотемпературной ползучести.
В рамках американской программы, направленной на создание сверхзвукового гражданского самолета, фирмами GE и PW по контрактам с NASA еще в 90-е годы был разработан легированный 3 % Ru и 6 % Re суперсплав четвертого поколения, получивший марку ЕРМ-102. Сплав используется или предполагается использовать для литья монокристаллических лопаток турбин двигателей F-11-129, CF6-80E, PW2037, PW4098, а также монокристаллических лопаток с проникающим охлаждением для двигателя JSF. Составы этого сплава и других известных зарубежных сплавов четвертого поколения: MC-NG (Франция), TMS-162 и TMS-196, приведены в табл. 1.
Таким образом, анализ представленных материалов указывает на то, что за рубежом (в Японии, США и Европе) решение проблемы создания газотурбинных двигателей для авиационной техники новых поколений тесно увязано с разработкой никелевых жаропрочных сплавов, легированных в первую очередь рением и рутением.
Разработчики последних отечественных сплавов ВЖМ-4 и ВЖМ-6 также продолжают следовать по пути повышения рабочих характеристик литейных никелевых жаропрочных сплавов легированием крайне дорогостоящими Re и Ru [2].
Таблица 1
Составы зарубежных монокристаллических никелевых
суперсплавов четвертого поколения
Сплав |
ρ, г/см3 |
Состав, % мас. |
||||||||||
Cr |
Со |
Мо |
W |
Ta |
Re |
Nb |
Al |
Ti |
Ru |
Другие |
||
ЕРМ 102 |
9,2 |
2,0 |
16,5 |
2,0 |
6,0 |
8,25 |
5,95 |
- |
5,55 |
- |
3,0 |
0,15Hf; 0,03С |
МС NC |
8,75 |
4,0 |
- |
1,0 |
5,0 |
5,0 |
4,0 |
- |
6,0 |
0,5 |
4,0 |
0,1Hf; 0,1Si, 0,03С |
TMS 162 |
9,04 |
2,9 |
5,8 |
3,9 |
5.8 |
5,6 |
4,9 |
- |
5,8 |
- |
6,0 |
0,1Hf; 0,03С |
TMS 196 |
9,02 |
5,4 |
6,0 |
1 |
1 |
6,2 |
6,4 |
0,5 |
5,5 |
- |
5 |
0,1Hf; 0,03С |
Данные направления развития суперсплавов не являются оптимальными по следующим причинам [3]:
- увеличение в сплавах содержания рения приводит к резкому удорожанию их стоимости. В настоящее время вследствие крайне малого объема мирового производства этого элемента (около 40 т в год) и активной политики США, скупающих Re по всему миру, его цена в течение трех последних лет выросла с 3,0 до 10,0 - 16,0 тыс. дол/1 кг. В результате стоимость жаропрочных никелевых сплавов последних поколений, содержащих рений и рутений, возросла с 3,0 млн. руб/т до 20,0 - 30,0 млн. руб./т. В тоже время стоимость современных газотурбинных двигателей составляет 1,5 - 2,0 тыс. дол/кг. Таким образом, впервые в мировой практике складывается ситуация, когда удельные цены жаропрочных сплавов становятся соизмеримыми с удельными ценами на газотурбинные двигатели, что с учетом интеллектуального и трудового вклада в создание и производство современных ГТД делает создавшуюся ситуацию достаточно проблематичной. Таким образом, крайне актуальной становится задача резкого снижения зависимости производимых в РФ никелевых жаропрочных суперсплавов от обеспеченности металлургического производства рениевой лигатурой, а также уменьшения их стоимости;
- легирование рением имеет не только положительную сторону, но и отрицательную. Этот элемент обладает эффективной способностью образовывать охрупчивающие сплав фазы, а его сильная ликвационная неоднородность и весьма низкая диффузионная подвижность усугубляют указанный эффект;
- совместное легирование сплавов Re и Ru также имеет определенные недостатки. Рутений - элемент платиновой группы, весьма дорого: его цена более 6,0 тыс. дол/кг. Он достаточно эффективно упрочняет сплав. Вместе с тем теоретические расчеты показывают, что влияние рутения на когезивную прочность матрицы никелевых сплавов почти в 2 раза ниже, например, вольфрама (W).
Указанное выше означает, что необходимо:
- иметь в РФ сплавы для лопаток ГТД, обладающие тем же, что и за рубежом, уровнем жаропрочных свойств, но при этом не имеющие в своем составе остродефицитный Re или легированные этим элементом в гораздо меньшем объеме по сравнению с зарубежными аналогами;
- разработать сплавы четвертого поколения, отвечающие по уровню эксплуатационных характеристик показателям зарубежных сплавов типа ЕРМ-102 (США), TMS-162 и TMS-196 (Япония) или превышающие их, но не имеющие в своем составе элемент платиновой группы - Ru.
В настоящее время в ОАО НПО «Сатурн» создан сплав СЛЖС-1, не имеющий в своем составе Re и Ru, однако уровень его высокотемпературной жаропрочности при 1000°С соответствует сплаву CMSX-4 (США), из которого изготавливаются монокристаллические лопатки ГТД истребителя F-22 пятого поколения [1]. Указанный сплав не имеет аналогов в мировой практике. В ОАО НПО «Сатурн» создан также монокристаллический сплав СЛЖС-3 четвертого поколения, обеспечивающий наиболее высокий среди известных в мире сплавов уровень жаропрочности при 1000°С, но при этом не имеющий в своем составе элемента платиновой группы - Ru (табл. 2).
Таким образом, успешно решаемая в настоящее время в России задача создания отечественных супержаропрочных никелевых сплавов обеспечивает:
- резкое снижение зависимости производства продукции оборонного назначения от остродефицитного рения;
- полное исключение использования дорогостоящих благородных металлов при изготовлении отечественных никелевых жаропрочных суперсплавов;
- уменьшение в несколько раз затрат на производство лопаток для эксплуатируемых и новых ГТД.
В таблице 2 представлены данные о перспективных никелевых суперсплавах с монокристаллической структурой для охлаждаемых лопаток ГТД третьего и четвертого поколений. Анализ результатов показывает, что сплав СЛЖС-3 обладает наиболее высокой жаропрочностью при 1000°С и испытаниях длительностью 100 и 300 ч. По параметру удельной длительной прочности этот сплав значительно лучше всех мировых аналогов, хотя стоимость его шихты в 1,4 - 2,0 раза ниже, чем у материалов-конкурентов.
Аналогичный уровень удельной длительной прочности имеет лишь последний сплав Японии TMS-196, однако цена его легирующих элементов является самой высокой из всех представленных в табл. 2 сплавов и вдвое превышает стоимость шихты сплава СЛЖС-3.
Проблема повышения температурной работоспособности монокристаллических сплавов требует обсуждения вопроса увеличения удельной массы (плотности) новых материалов [1].
Дело в том, что прочность сплава при высоких температурах обеспечивают легирующие элементы, имеющие наиболее высокую температуру плавления и наименьшую диффузионную подвижность, контролирующую процессы структурных изменений в материале. К этим элементам в первую очередь относятся вольфрам, тантал и рений, которые имеют весьма высокую удельную массу и легирование ими приводит (наряду с заметным увеличением жаропрочности сплавов) к росту плотности.
Таблица 2
Перспективные никелевые суперсплавы с монокристаллической структурой
Сплав, страна |
Особенности легирования |
Свойства при 20°C |
Свойства при 1000 °C |
Удельная длительная прочность, σ300ч1000°С/d, *10-3,см |
|||
σв МПа |
σ0,2 МПа |
δ % |
σ100 МПа |
σ1000,МПа (τ=300 ч) |
|||
СЛЖС-3, Россия |
4,8%Re, |
1170 |
980 |
8,0 |
347 |
300 |
31,8 |
TMS-196, Япония |
6,4%Re, 5%Ru |
- |
- |
- |
328 |
240 |
32,0 |
TMS-162, Япония |
5%Re, 6%Ru |
- |
- |
- |
320 |
230 |
31,0 |
ЕРМ-102, США |
- |
- |
- |
- |
325 |
200 |
28,9 |
ВЖМ-1, Россия |
9%Re |
1190 |
945 |
21 |
330 |
215 |
31,2 |
ВЖМ-4, Россия |
6,5%Re, 4%Ru |
1220 |
865 |
20 |
315 |
200 |
28,7 |
ВЖМ-6, Россия |
Re+Ru=11% |
- |
- |
- |
- |
220 |
30,7 |
MC-NG, Франция |
4%Re, 4%Ru |
- |
- |
- |
275 |
190 |
26,8 |
CMSX-10 (RR 3000), США-Великобритания |
6%Re |
1190 |
985 |
21 |
290 |
185 |
27,2 |
Таким образом, увеличение жаропрочности никелевых сплавов связано с ростом удельной массы. Анализ данных по никелевым монокристаллическим сплавам от первого до четвертого поколений показывает, что если среднее значение удельной массы сплавов первого поколения составило 8,45 г/см3, то второго поколения 8,8 г/см3, третьего поколения 9,0 г/см3 и четвертого поколения 9,03 г/см3 [1, 3]. Однако при этом их удельная жаропрочность растет гораздо более высокими темпами, обеспечивая успешную эксплуатацию новых сплавов в газотурбинных двигателях. В частности, удельная жаропрочность (при длительности испытаний 300 ч) монокристаллических сплавов первого поколения CMSX-2 и ЖС-40 составила 20,9 и 22,6 соответственно, в то время как этот же показатель для монокристаллических сплавов четвертого поколения составляет величину 30-32 (см. таблицу 2), т. е. за 30 лет был совершен скачок примерно в 1,5 раза, что является весьма высоким показателем роста жаропрочности никелевых сплавов за все время их эксплуатации в ГТД.
Сплавы СЛЖС-3 и СЛЖС-1 имеют достаточно высокие значения удельной массы соответственно 9,4 и 9,15 г/ см3. Однако при этом они обладают наибольшими (для своего класса) параметрами удельной длительной прочности.
Пример этому: в свое время сплав ЖС-32 также имел наибольшую удельную массу среди сплавов-аналогов (8,8 г/см3) , отличаясь существенно более высокими показателями жаропрочности и удельной длительной прочности. Именно это обстоятельство побудило Генеральных конструкторов A.M. Люльку, С.П. Изотова и др. применять его в своих новых двигателях, и даже спустя более чем 25 лет этот сплав является одним из наиболее надежных и востребованных промышленностью.
Таким образом, анализ представленных результатов указывает на возможность создания никелевых жаропрочных сплавов с монокристаллической структурой, имеющих современный уровень жаропрочности, но при этом содержащих в значительно меньшем количестве остродефицитные и крайне дорогостоящие элементы - рений и рутений, или не имеющих таковых вообще в своем составе.
Предложенные сплавы СЛЖС-1 и СЛЖС-3 могут быть использованы в перспективных газотурбинных двигателях и одновременно (обладая более высокой удельной жаропрочностью и в несколько раз более низкой стоимостью) с успехом применяться в уже существующих ГТД (в частности, вместо широко используемого сплава ЖС-32ВИ).
Литература
1. Логунов А.В., Шмотин Ю.Н. Тенденции разработки и применения Ni-суперсплавов для лопаток ГТД в современных и перспективных силовых установках авиационного назначения// Технология легких сплавов. – 2011. - №4. – С. 11-17
2. Логунов А.В., Розумовский И.Н., Ларионов В.Н. и др. Жаропрочные никелевые сплавы, получаемые методом монокристального литья, для деталей перспективных двигателей//Перспективные материалы. – 2008. - №3. – С. 1-9
3. Логунов А.В., Шмотин Ю.Н., Маринин С.Ф. и др. Теоретические основы газостатической обработки литых деталей с монокристаллической структурой и жаропрочных никелевых сплавов//Вестник МГОУ. Сер. «Техника и технология». – 2010. - №2.- С. 27-35
ВУНЦ ВВС «ВВА»
УДК 541.1: 546.171. 001.24
В.П. Горшунова, В.В. Пусовская, И. А. Ходякова
ВЗРЫВОПОЖАРООПАСНЫЕ СВОЙСТВА АММИАКА И ИХ ОЦЕНКА НА ОСНОВЕ ТЕРМОДИНАМИЧЕСКИХ РАСЧЕТОВ
На основе термодинамических расчетов в двух приближениях – по методам Улиха и Темкина-Шварцмана, произведена оценка взрывопожаро-опасных свойств аммиака
Смесь паров NH3 c воздухом при объемном содержании аммиака от 15 до 28% (107…200мг/мл) является взрывоопасной. Давление взрыва аммиачно-воздушной смеси может достигнуть 0, 45 мПа при объемном содержании аммиака в воздухе свыше 11% (78,5 мг/л). При наличии открытого пламени начинается горение NH3. Температура воспламенения по разным литературным источникам 630 – 650 оС [1,2]. В присутствии катализатора можно достигнуть сгорания смесей с воздухом уже при относительно низких температурах (300 – 500 оС). В этих условиях образуются оксиды азота [2].
Аммиачно-воздушные смеси характеризуются большими значениями минимальной энергии зажигания (680 мДж), благодаря чему существуют ограниченные возможности воспламенения аварийных выбросов аммиака в открытую атмосферу и производственные помещения. Однако известно немало случаев, когда на производствах, использующих большие количества аммиака, происходили взрывы аммиачно-воздушных смесей.
В качестве примера подобной аварии можно привести происшествие на ПО «АОЗТ» (г.Ионова, Литва). Здесь 20.03.93 г. рухнул резервуар с 7000 т аммиака. Начался пожар, заражение воздуха оказалось значительным, погибло 7 человек, пострадало 50. Всего из опасной зоны было эвакуировано около 30 тыс. человек. В атмосфере возникла значительная концентрация оксида азота (II), который является сильным ядом, поражающим кровь [3].
В России 28.04. 2004 г. на Очаковском хладокомбинате произошел взрыв аммиака в результате его утечки из трубопровода холодильной установки. Масса аммиака составила 40 кг. В результате взрыва обрушилась стена площадью 300 м2. Общая площадь разрушения достигла 400 м2. В эпицентре взрыва концентрация аммиака в 50 раз превысила установленные нормативы и составила 1000мг/м3, в то время как ПДК для аммиака составляет 20 мг/м3. Зданию хладокомбината нанесен значительный материальный ущерб. В ликвидации аварии принимали участие свыше 300 спасателей и пожарных, в том числе специальные отряды противохимической защиты. О числе пострадавших не сообщается.
Целью данной работы явилось обоснование пожаровзрывоопасных свойств аммиака на основе термодинамических расчетов. Известно, что в основе горения аммиака лежит химическая реакция:
NH3 + 3/4O2 = 1/2N2 + 3/2H2O. (1)
Чтобы оценить химическое сродство реагентов, т.е. аммиака и кислорода, необходимо рассчитать Gх..р. при температуре воспламенения – 650 оС (950К). Были использованы: приближенный расчет по методу Улиха и точный расчет по методу Темкина – Шварцмана [4].
Согласно первому приближению (метод Улиха) предполагается, что тепловой эффект и энтропия химической реакции (1) не зависят от температуры, и константа равновесия может быть рассчитана по формулам:
R
ln
KT
= -
![]()
или
lnKT
= -
![]() .
(2)
.
(2)
Н![]() рассчитывали на основании первого
следствия из закона Гесса по формуле
рассчитывали на основании первого
следствия из закона Гесса по формуле
Н
=![]() nН
nН![]() (прод.)
-
nН
(исх.).
(3)
(прод.)
-
nН
(исх.).
(3)
S находили из соотношения
S
=
![]()
![]() (4)
(4)
В выражениях (3) и (4) расчеты термодинамических функций производили с учетом стехиометрических коэффициентов в уравнении реакции, а их стандартные значения находили в справочниках термодинамических величин [5,6].
Рассчитанная в первом приближении константа равновесия реакции (1) при температуре 950К оказалась равной К950 = 1,261019. G950 находили, используя известное соотношение
G950 = - RT lnK950. (5)
Можно применить выражение
G950 = H - TS , (6)
где Т равна 950К. Получили приближенное значение G950 = -346,72 кДж.
Согласно второму приближению (методу Темкина-Шварцмана) учитывается зависимость теплового эффекта химической реакции от температуры в соответствии с законом Кирхгоффа. В этом случае используется температурная зависимость теплоемкости в виде эмпирических степенных рядов. Выражение для расчета К950 имеет вид:
RlnK950
-![]() +
+
+а2![]()
![]() .
(7)
.
(7)
Значения
коэффициентов
![]() находили
в справочнике термодинамических величин
[6]. Константа равновесия данной реакции
при 950К оказалась равной К950=
1,811014,
а G
находили
в справочнике термодинамических величин
[6]. Константа равновесия данной реакции
при 950К оказалась равной К950=
1,811014,
а G![]() ,
рассчитанное по формулам (5) или (6), равно
– 350,04 кДж.
,
рассчитанное по формулам (5) или (6), равно
– 350,04 кДж.
Если реакция окисления аммиака происходит при соприкосновении с металлом, способным оказать каталитическое действие на реакцию, то продуктами взаимодействия будут пары воды и оксид азота (II), как это было в случае аварии на производстве в г. Ионова:
NH3 + 5/4O2 = NO + 3/2H2O . (8)
В
соответствии с приближенным расчетом,
выполненным по формулам (2), (3), (4), значение
константы равновесия реакции (8) К![]() равно
6,281014,
а
равно
6,281014,
а
![]() оказалось равным –269,2 кДж. Найденная
величина константы равновесия последней
реакции по методу Темкина-Шварцмана в
соответствии с выражением (7) равна
9,251014,
а величина G
-
(-272,0) кДж.
оказалось равным –269,2 кДж. Найденная
величина константы равновесия последней
реакции по методу Темкина-Шварцмана в
соответствии с выражением (7) равна
9,251014,
а величина G
-
(-272,0) кДж.
Полученные термодинамические константы интересно сравнить с соответствующими константами реакции взаимодействия водорода с кислородом, являющейся классическим примером разветвляющейся цепной реакции. Ее протекание можно выразить следующим суммарным уравнением реакции:
Н2 + 1/2O2 = H2O. (9)
Приближенный расчет по Улиху дал значение К850, равное 3,651012, а G850, рассчитанное по формуле (5), оказалось равным –204,1 кДж. Точный расчет с использованием формул (5), (6) и (7) дал следующие результаты: К850 = 9,921012 и G850 = -207,95 кДж.
Данные расчеты выполнены для температуры 850К, так как в соответствии с литературными источниками [2], температура воспламенения стехиометрической смеси газов водорода и кислорода в соотношении, соответствующем уравнению (9) («гремучий газ»), лежит при температурах около 500о С.
Известно [7], что цепной взрыв, или воспламенение, наблюдается при протекании цепных реакций с разветвленными цепями, к которым и относится реакция взаимодействия водорода с кислородом. Эти реакции характеризуются верхним и нижним пределами воспламенения, которые зависят от температуры, формы сосуда, содержания примесей в газовой смеси. Так, для водородно-кислородных смесей при 298К и общем давлении 101,3 кПа нижний предел воспламенения составляет около 6 об.% кислорода, верхний предел – около 95 об.% кислорода. Для полного сгорания 1 объема водорода требуется около 2,4 объемов воздуха. Чистый гремучий газ взрывается, начиная с 500 оС. В присутствии некоторых катализаторов взаимодействие происходит уже при значительно более низкой температуре.
Приведенные факты говорят о большом сходстве в протекании реакций окисления, идущих по цепному механизму. Рассмотренные выше процессы с участием аммиака (1,8) также можно отнести к этой группе реакций. Термодинамический анализ подтверждает данное высказывание. Действительно, значения констант равновесия реакций окисления аммиака и водорода при соответствующих температурах воспламенения имеют большую величину: 1014 – 1019 для реакций окисления аммиака и 1012 – для реакции окисления водорода. Кроме того, величины химического сродства G при температурах воспламенения одного порядка: для реакций окисления аммиака -350 и -270 кДж, а для реакции окисления водорода - 208 кДж. Термодинамические расчеты показывают, что при температуре воспламенения реакции взаимодействия аммиака с кислородом протекают даже с меньшей величиной энергии Гиббса, а значит с большим химическим сродством, чем реакция окисления водорода. Следовательно, это может служить косвенным доказательством того, что подобно «гремучему газу» смесь аммиака с кислородом (а также с воздухом) является пожаровзрывоопасной.
Литература
1. Вредные вещества: Справочник / Под ред. Н.В. Лазарева.-М.: Химия, 1971. – 365 с.
2. Реми Г. Курс неорганической химии. – М.: Мир, 1972 – 1974. – 914 с.
3. Гринин А.С., Новиков В.Н. Экологическая безопасность. Защита территории и населения при чрезвычайных ситуациях: Учеб. пособие. - М.:ФАИР-ПРЕСС, 2000.-336 с.
4. Киреев В.А. Методы практических расчетов в термодинамике химических реакций. - М.: Химия. 1975. – 536 с.
5. Термодинамические свойства индивидуальных веществ: Справочное издание. – М.: Наука, 1978 – 1983 . –767 с.
6. Краткий справочник физико-химических величин / Под ред. А.А. Равделя, А.М. Пономаревой. – Л.: Химия, 1993. – 331 с.
7. Стромберг А.Г. Физическая химия. – М.: Высш. школа. 2003. – 527 с.
Воронежский государственный технический университет
УДК 621.579
Н.А. Поленова, Б.А. Спиридонов
ПОЛУЧЕНИЕ НАНОСТРУКТУРИРОВАННОЙ АЛЮМИНИЕВОЙ ФОЛЬГИ, ИСПОЛЬЗУЕМОЙ В ПРОИЗВОДСТВЕ КОНДЕНСАТОРОВ
Рассмотрены условия получения нанопористого оксида алюминия (ПОА) и его некоторые свойства. Показаны перспективы применения анодированной алюминиевой фольги, используемой в производстве конденсаторов
Известно, что в производстве алюминиевых электролитических конденсаторов используется Al фольга высокой чистоты (99,99 %) толщиной 50, 100 и 200 мкм. На начальном этапе алюминиевая фольга подвергается травлению для увеличения ее удельной поверхности при заданной площади анода. При шероховатой поверхности анода (в результате травления) созданный при формовке тонкий оксидный слой следует за всеми неровностями электролит, который служит второй обкладкой, также заполняет неровности поверхности, что позволяет заметно увеличить емкость, приходящуюся на единицу площади по сравнению с гладким, нетравленым анодом. Отношение общей поверхности травленого анода Sпов. К его площади SA носит название коэффициента травления: КТР.= Sпов. / SA . Коэффициент травления снижается с увеличением формовочного напряжения Uф., т.е. с увеличением толщины оксидного слоя. Если при малых Uф. можно получить КТР. ≈ 10-20, то при Uф. порядка сотен вольт КТР. снижается до 5-10. Это объясняется тем, что при повышенной толщине оксида уже не будет сказываться влияние наиболее мелких неровностей на поверхности анода [1-4].
Ранее применялось химическое травление алюминиевой фольги в растворах HCl, HCl с добавкой CuCl2 или смеси HCl с HNO3 при повышенных температурах (70 – 90 0С).
В настоящее время основным методом является электрохимическое травление в растворе NaCl при температуре 40 – 60 0С, при котором на анодную фольгу, проходящую через травильную ванну, подается небольшой положительный потенциал, создающий плотность тока порядка 0,75 – 0,15 A/см2 . Активирующее действие на анодное растворение алюминия оказывают ионы хлора (l-1), которые разрушают защитную оксидную пленку. Электрохимическое позволяет не только удешевить травящий раствор, но и уменьшить его вредность. Изменением плотности тока в травильной ванне можно регулировать процесс травления, что также является преимуществом этого метода; он позволяет также получать большую однородность значений КТР. По длине фольги.
Для фольги 99,992 – 99,995 % Al можно получать КТР = 8 ± 0,8 при Uф. = 480 В и 13 ± 1,3 при Uф. = 10 В. Фольгу более высокой чистоты получают методом зонной плави.
В последние годы с целью существенного увеличения емкости конденсаторов используют наноструктурированную алюминиевую фольгу, которую получают по различным технологиям.
Экспериментально установлено [5], что пористые пленки анодного оксида алюминия являются примером самоорганизующихся структур, параметры которых можно представить в виде гексагональной упаковки цилиндрических пор, расположенных строго перпендикулярно плоскости пленки. Уникальная пористая структура анодированного алюминия, параметры которой можно варьировать в процессе синтеза, позволяет использовать высокоупорядоченные пленки пористого оксида алюминия (ПОА) в качестве матриц для создания нанокомпозитов [6].
Электролиты для анодного окисления алюминия принято разделять на две группы:
1) Электролиты, в которых оксид алюминия практически нерастворим. Образующаяся пленка почти не имеет пор и, являясь диэлектриком, уже в очень тонком слое препятствует прохождению электрического тока. Полученные таким способом пленки тонкие, менее 1 мкм, но плотные и относятся к пленкам барьерного типа. Электролиты этого типа представляют собой обычно растворы слабых неорганических и органических кислот, например, борную, винную, лимонную или их солей. Толщина пленки (d, нм) пропорциональна напряжению (U, В) на электролизере:
D = a · U (1)
где а – коэффициент пропорциональности, приблизительно равный 1,4 × 10-7 см/В.
2). Электролиты, в которых оксид алюминия достаточно хорошо растворим. Благодаря такой способности образуется пористая и проницаемая для электролита пленка; она может достигать значительных толщин, которые в зависимости от состава раствора и режимов электролиза находятся в интервале от 1 до 500 мкм. К электролитам этого типа принадлежат растворы хромовой, щавелевой, сульфосалициловой кислот и некоторые другие более сложного состава.
Образующаяся оксидная пленка состоит из двух слоев: тонкого (от 0,01 до 0,1 мкм) барьерного беспористого слоя и более толстого пористого. Механизм возникновения и роста анодной пленки еще недостаточно изучен, поскольку представляет собой совокупность сложных физико-химических явлений. Н.Д. Томашовым было предложено, что тонкий барьерный слой образуется и растет в результате взаимодействия ионов Al3+ и кислорода при их встречной миграции в барьерном слое электрического поля большей напряженности. Процесс анодирования протекает в три стадии:
1) передача кислорода от анионов или молекул раствора на анодируемый металл и возникновение и возникновение первичного соединения алюминия с кислородом;
2) формирование сплошной тонкой оксидной пленки барьерного типа;
3) рост утолщенной пористой пленки.
Донором кислорода при реализации такого механизма являются молекулы воды и кислородсодержащие анионы электролита (SO42-, HSO4-, PO43-, Cr2O42- и др.). Реакция анодного окисления алюминия в общем виде протекает по схеме:
2Al + 3H2O → Al2O3 + 6H+ + 6ẽ (2)
Эта реакция является суммарной ряда частных процессов, наиболее вероятный из которых – анодная ионизация алюминия на границе раздела металл – барьерная оксидная пленка:
Al – 3e- → Al3+ (3)
Выделяющиеся по этой реакции электроны уходят во внешнюю цепь, образуя ток анодирования. Ионы Al3+ мигрируют в электрическом поле оксида (в его ионной решетке) к внешней поверхности барьерного слоя. Поскольку при анодировании алюминия заметного выделения кислорода не происходит, процесс его передачи протекает на границе раздела барьерный слой – электролит, формируя анодный процесс образования ионов кислорода: H2O → 2H+ + О2-. (4)
Ионы кислорода, образующиеся на поверхности барьерного слоя, могут мигрировать в нем под действием электрического поля по направлению к металлу навстречу ионам Al3+.
Образование пор начинается в энергетически выгодных местах (границы зерен, линии скольжения дислокаций, структурные дефекты и др.). Первоначально поры на внешней поверхности распределяются случайно, хаотично, неупорядочено. В процессе их роста на внешней границе металл – оксид одновременно начинает расти новый слой оксида, отдельные ячейки которого имеют вид полусферы.
С увеличением напряжения на электролизере между имеющимися ячейками образуются новые, которые постепенно заполняют всю поверхность металла. После того, как отдельные ячейки сомкнутся и покроют всю поверхность металла, начинается рост каждой ячейки в глубину с последующим формированием каналов. Поскольку ячейки, имеющие меньшую полусферу в основании, характеризуются меньшей толщиной барьерного слоя, то именно они будут иметь большую возможность для роста, что и проводит к образованию ячеистой упорядоченной структуры.
Механизм роста и растворения оксида был предложен Гоаром и Яколом, в соответствии с которым этот процесс обусловлен увеличение поля на дне пор. Возникновение пор происходит не сразу, а только после достижения некоторой критической толщины барьерного слоя на дне поры, при котором становится возможным проникновение в оксид протона, участвующего в реакции растворения против анодного поля.
Исследованиями было установлено, что и барьерные и пористые пленки, развивают ячеистую структуру только после истечения минимального времени электролиза (τ), при котором ток имеет также низкие значения в режиме постоянного напряжения. В порообразующих электролитах это время составляет примерно 2 секунды, в 3% растворе тартрата аммония (рН = 5,5), τ = 12 мин, а в том же электролите, но при рН = 7,7 τ = 90 мин. В порообразующих электролитах минимум плотности тока соответствует началу формирования пор. Последующее увеличение тока (при постоянном формирующем напряжении) отражает утонение барьерного слоя на дне пор по мере их роста. Дальнейшая стабильность тока означает стабильность роста барьерной пленки (с постоянной анодирования ~ 1,0 нм/В) и поры растут преимущественно в пленке, но не за счет подложки (металла). По мере роста большие поры поглощают соседние более мелкие, образуя новые поры. Таким образом, происходит распределение пор по размерам и формам. Только после некоторого времени диаметр пор становится постоянным, до этого момента он подчиняется распределению:
![]() (5)
(5)
где d – диаметр пор; h – толщина барьерного слоя; τ – время анодирования пор; τ0 – время зарождения пор; К – постоянная анодирования.
Полная застройка оксидными ячейками поверхности металла происходит через несколько секунд (4 – 7 с) и в дальнейшем структура пленки не изменяется.
На второй стадии происходит упорядочение ячеисто-пористой структуры путем продолжения анодирования участков алюминия в тангенциальном направлении (между растущими оксидными ячейками). Замедления растворения объясняется снижением количества инжектируемых ионов алюминия.
Поскольку электрическое поле около единичной поры стремиться быть сферическим, то передний фронт передней растущей оксидной пленки также будет сферическим, если пора действительно является точечным источником. Но так как пора имеет конечный размер, передний фронт ячейки будет иметь форму сектора. После длительного анодирования и слияния отдельных ячеек образуются ячейки с цилиндрическими порами в центре и с промежуточными, сквозными, металлическими столбиками с треугольным сечением (рис. 1).
Остатки металла между ячейками еще находятся в действующей электрической цепи, и поэтому будут превращаться в оксид анодным способом до окончательного слияния ячеек. Металл при этом расходуется в равной степени с каждой стороны под влиянием тока в порах трех окружающих ячеек. Когда весь металл из этих столбиков израсходуется полностью, слой оксида станет непрерывным, и ячейки приобретут форму гексагональных призм, а не цилиндров.
Рис.1. Схематическое изображение фрагмента пористого оксида с плотно упакованными гексагональными ячейками в центре с порой. D – диаметр ячейки, h – высота поры
Анализ теоретических и экспериментальных результатов позволяет определить последовательность процессов при формировании самоупорядоченной структуры пористого оксида алюминия (рис. 2):

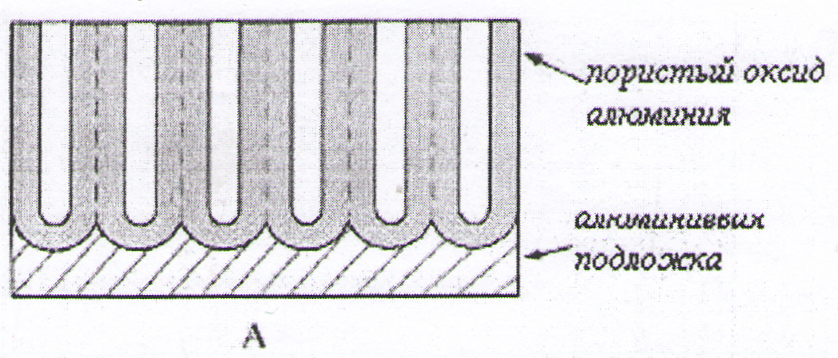
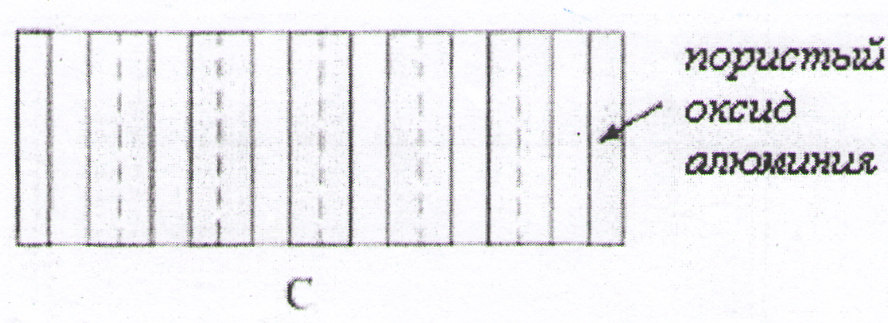
Рис. 2. Процесс получения ПОА: А-формирование ПОА на алюминиевой фольге; В-отделение ПОА от остатков алюминиевой фольги; С - удаление барьерного слоя
Этапы формирования ПОА:
-образование беспористой пленки барьерного типа;
- зарождение пор, хаотично распределенных по поверхности;
- развитие пор и ячеек на границе раздела оксид-металл в барьерном слое;
- формирование регулярных ячеек и пор цилиндрической формы с постоянным диаметром за счет подложки (рост в глубину);
- формирование гексагонально-упорядоченных ячеек пористого оксида.
Процесс самоупорядочения при росте пористого оксида наиболее наглядно проявляется на последней самой длительной стадии процесса (именно она является лимитирующей). При этом образуются оксиды большей толщины, которая определяется временем анодирования.
При проведении анодирования при низких температурах (около 0 ºС) и циркуляцией электролита, длительном времени электролиза (до 160 ч), возможно сформировать регулярную периодическую структуру пористого оксида алюминия.
Сравнительно простой способ синтеза пористого оксида алюминия из толстых подложек металла показан в работе. Он основан на предварительном выращивании «жертвенного» пористого оксида толщиной порядка 100 мкм. Показано, что по мере увеличения толщины растущего оксида случайное расположение пор преобразуется в упорядоченную структуру. После селективного удаления «жертвенного» слоя оксида поверхность алюминия наследует упорядоченный рельеф предыдущего слоя. Последующее анодирование с таим рельефом, приводит к формированию оксида с высокой степенью упорядоченности.
По одному из методов первая стадия осуществляется из раствора щавелевой кислоты при 40 В и 15 ºС в течение 12 часов, после чего смесью кислот фосфорной и хромовой активируется поверхность алюминия. Во втором процессе анодирования участвует упорядоченная вогнутая структура на нижнем слое металла. После второго анодирования оставшийся нижний слой алюминия удаляется в растворе хлорида меди при 15 ºС в течение 3 часов. Дно пор последовательно открывается в 0,1 М растворе фосфорной кислоты при 35 ºС. Затем образец погружают в ацетон и промывают в течении 15 мин, получая в результате чистую пористую пленку.


Рис. 3. Изображение ПОА: а) на первой; б) завершающей стадии его формирования, полученное на СТМ
С целью получения пленок оксида алюминия с высокой степенью упорядоченности разработан способ анодирования алюминия, основанный на динамическом изменении во времени температуры зоны электрохимической реакции. Анодирование алюминиевой фольги проводили в растворах серной, щавелевой и ортофосфорной кислот в гальваностатическом или потенциостатических режимах. Было высказано предположение, что наиболее эффективное упорядочение оксида будет происходит при фиксированном во времени размере пор. Это условие можно представить в виде:
![]() (6)
(6)
где Dp – диаметр поры (нм); j – плотность тока (мА/см2);
Т – температура электролита, К; a, b, c, γ – эмпирические коэффициенты, которые могут быть определены для каждого электролита.
Из выражения (6) следует, что зависимость температуры зоны реакции от плотности электрического тока в электрохимической ячейке – линейна. На основании экспериментальных данных было установлено, что сохраняя оптимальное соотношение между Т и j, можно обеспечить минимальное изменение диаметра поры при постоянном периоде структуры оксида, тем самым создать условия для выращивания анодного оксида с повышенной степенью упорядоченности структуры.
Режимы получения ПОА представлены в таблице.
Режимы получения ПОА

Установлено, что максимальные плотности тока стационарного гальваностатического анодирования алюминия ограничиваются природой и концентрацией электролита (не более 10 – 15 мА/см2). Процесс анодирования необходимо проводить при перемешивании и температуре ниже комнатной, так как при более высокой температуре скорость растворения оксида алюминия становится больше скорости его образования. В меньшей степени на скорость образования пористого оксида оказывает температура при проведении анодирования в сульфосалициловой кислоте. По морфологическим параметрам пористый оксид алюминия, получаемый в сульфосалициловой кислоте (ССК) занимает промежуточное положение между оксидами, получаемыми анодированием в серной и щавелевой кислотах. В водном растворе ССК с концентрацией 0,6 и 1 М скорость гальваностатического анодирования возрастает пропорционально плотности анодного тока.
Установлено, что с повышением плотности тока от 1 до 400 мА/см2 коэффициент объемного роста увеличивается и принимает значения от 1,26 до 1,63 и практически не зависит от концентрации кислоты. Было также выявлено, что при значениях анодной плотности тока 30 – 40 мА/см2 коэффициент объемного роста имеет максимум.
Дальнейшие исследования позволили установить, что независимо от природы электролита растворение барьерного слоя начинается одновременно с его образованием. Было выявлено, что с увеличением скорости подъема анодного потенциала наблюдается заметное возрастание анодного тока и ускорение процесса растворения в основном за счет перехода ионов Al3+ из оксида в электролит.
В
последние годы получили развитие новые
нанотехнологии для получения наноматериалов
с уникальными характеристиками, например,
для производства долгоживущих
конденсаторов очень высокой емкости,
стабильно работающих от -60 до +80 0С.
Они просто незаменимы там, где перепады
температуры значительны, например, в
авиации. В этих конденсаторах
В качестве примера можно остановиться на технологии получения конденсаторной фольги из специального алюминиевого проката с кубической структурой. После направленного травления получаются в объеме фольги туннели в диаметре от нескольких десятков нанометров и длиной до нескольких сотен нанометров. В последующем оксидируют стенки туннелей оксидами со специальной структурой. Толщина оксидов определяется напряжением оксидирования и составляет от 10-12 нм. И выше. Специальными методами пропитывают эти оксидированные туннели электролитом. В результате получается структура: металл – оксид – электролит (МОЭ). В ионисторах используют эффект двойного электрического поля в месте контакта углеродного слоя с ионами электролита. При этом величина пор углеродного слоя электрода сравнима с диаметром иона электролита и составляет в среднем 2-4 нм.
Суперконденсаторы (ионисторы) относятся к классу конденсаторов использующих энергию заряда сосредоточенного в двойном слое. Напряжение одного суперконденсатора составляет 1,8-3 В, номинальная емкость до 10000 Ф., вес до 1 кг., габарит 95 мм ´ 120 мм (61 мм ´ 61 мм ´160 мм). В процессе эксплуатации и хранения ионисторы не требуют обслуживания, имеют длительный срок эксплуатации. Энергия запасаемая ионисторами может достигать 50-60 дж/г, а мощность десятки кВ/кг.


Рис.4. Фольга с двойным электрическим (углеродным) слоем
Суперконденсаторы (ионисторы) относятся к классу конденсаторов использующих энергию заряда сосредоточенного в двойном слое. Напряжение одного суперконденсатора составляет 1,8-3 В, номинальная емкость до 10000 Ф., вес до 1 кг., габарит 95 мм ´ 120 мм (61 мм ´ 61 мм ´160 мм). В процессе эксплуатации и хранения ионисторы не требуют обслуживания, имеют длительный срок эксплуатации. Энергия запасаемая ионисторами может достигать 50-60 Дж/г, а мощность десятки кВ/кг.
В последние годы разработаны ионисторы с применением электродов с угольной тканью (рис. 4). Основная область применения суперконденсатров (ионисторов) – гибридная энергетическая установка новых транспортных средств, обладающих высокими эксплуатационными и экологическими характеристиками. Ионисторы, которые обладают высокой мощьностью и коротким временем заряда используются в системах электростартерного пуска двигателей автомобилей и тракторов, дизельных двигателей судов и тепловозов и других энергетических установок.
Литература
1. Рене В.Т. Электрические конденсаторы. Л.: Энергия, 1969. 602 с.
2. Кучинский г.С., Назаров Н.И. Силовые электрические конденсаторы. М. : Энергия, 1975. 256 с.
Меркулов В.И. Основы конденсаторостроения. Учебн. Пособие. Томск. Изд.- во ТПИ. 2001. 121 с.
Меркулов В.И. Основы конденсаторостроения. Лаборат. Практикум. Изд.-во ТПИ, 2011. 84 с.
Пул Ч., Оуэнс Ф. Нанотехнологии. М.: Техносфера. 2006.- 336 с.
Белов А.Н., Гаврилов С.А., Шевяков В.И. Особенности получения наноструктурированного анодного оксида алюминия. Российские технологии. 2006.- Т. 1. № 1-2. С. 223-227.
Воронежский государственный технический университет
УДК 621.892.5
К.А. Журавский, А.В. Петров
ПЕРСПЕКТИВНЫЕ МЕТОДЫ НАНЕСЕНИЯ РАДИОПОГЛОЩАЮЩИХ МАТЕРИАЛОВ
В статье рассматривается перспективный метод дифференцированного распределения радиопоглощающих свойств материалов по объему покрытия с целью уменьшения эффективной площади рассеяния
Необходимость разработки радиопоглощающих материалов для создания малозаметных самолетов стала логическим следствием развития системы ПВО, основанная на радиоэлектронных средствах обнаружения и наведения оружия. И сразу возникла задача разработки самолетов с низкой эффективной площадью рассеяния (ЭПР).
Реальными плодами этих разработок стало создание ряда самолетов «Стелс» с малой заметностью. В фирме Локхид такой самолет был создан в 1976 году.
Согласно американской терминологии, понятие термина «Стелс» включает уменьшение всех возможных демаскирующих факторов, а также включает активные и пассивные средства (устранение эффективных отражателей радиолокационных волн) противодействия обнаружению.
Основные особенности самолета с малой ЭПР: внутреннее размещение двигателей, плавное сопряжение крыла с фюзеляжем, минимальные площади боковых и фронтальных поверхностей. Из-за объема, занимаемого двигателями и воздухозаборниками, выхлопными системами и оружием, размещенными внутри фюзеляжа, самолет «Стелс» требует больших внутренних объемов по сравнению с обычными самолетами тех же размеров.
Важным фактором, обеспечивающим малую ЭПР, является применение радиопоглощающих материалов (РМ), преобразующих энергию радиолокационного излучения в тепловую энергию в результате взаимодействия с электромагнитным излучением.
РМ подразделяют на широкополосные, узкополосные, комбинацию двух типов и радиопоглощающие краски.
К первому типу относятся углеродистые соединения. Их применение наиболее целесообразно в специально разработанных поглощающих конструкциях. Например, в конструкциях сотового типа радиопоглощающий пеноматериал можно наносить на стенки сот. Такие сотовые конструкции можно применять для изготовления обшивки самолета. Разработанные в начале 60х годов порошковые РМ обеспечивали поглощение 95% энергии попадающего на них радиолокационного излучения в диапазоне 3,5-13,0 ГГц при толщине материала 25 мм. Приблизительно такими же радиопоглощающими свойствами обладает и материал кевлар. Углеродосодержащие РМ предполагается использовать в выхлопных системах для обеспечения малой ЭПР силовых установок. Одним из способов применения РМ является их использование для подавления отражения радиолокационных волн от угловых стыков и острых кромок. Однако, для обеспечения необходимой эффективности покрытия, эти материалы должны иметь большую толщину и, вероятно, не пригодны для нанесения на металлическую обшивку самолета.
Узкополосные РМ применяют в многослойных покрытиях, внутренняя поверхность которых образована металлическим отражающим слоем. Такие покрытия настроены на определенную длину РЛС противника. Оптимальная толщина покрытия соответствует 1/4 длины волны РЛС. Подобные покрытия используют также для генерации равноамплитудного отраженного сигнала, нейтрализующего основной отраженный сигнал /1/.
Поглощающие покрытия содержат специальные поглощающие компоненты (графит, порошки ферритов и пр.), а структура покрытий обеспечивает снижение отражений от границы раздела сред (воздух — покрытие). Различие между собственно материалами (РПМ) и покрытиями (РПП) до некоторой степени условно и предполагает, что первые входят в состав конструкции объекта, а вторые — как правило, наносятся на его поверхности. Условность разделения связана и с тем обстоятельством, что любой радиопоглощающий материал является не только материалом, но микроволновым устройством-поглотителем. Способность материала поглощать высокочастотное излучение зависит от его состава и структуры. РПМ и РПП не обеспечивают поглощения излучения любой частоты, напротив, материал определенного состава характеризуется лучшей поглощающей способностью при определенных частотах. Не существует универсального поглощающего материала, приспособленного для поглощения излучения радиолокационной станции (РЛС) во всем частотном диапазоне.
Существует, по меньшей мере три типа РПМ: резонансные, нерезонансные магнитные и нерезонансные объёмные материалы. Резонансными или частотнонастроенными РПМ обеспечивается частичная или полная нейтрализация отраженного от поверхности поглотителя излучения частью его, прошедшей по толщине материала. Эффект нейтрализации значителен при толщине поглотителя, равной одной четверти длины волны излучения. В этом случае, отраженные поверхностью поглотителя волны находятся «в противофазе».
Резонансные материалы наносятся на отражающие поверхности объекта маскировки. Толщина РПМ соответствует четверти длины волны излучения РЛС. Падающая энергия высокочастотного излучения отражается от внешней и внутренней поверхностей РПМ с образованием интерференционной картины нейтрализации исходной волны. В результате происходит подавление падающего излучения. Отклонение ожидаемой частоты излучения от расчётной приводит к ухудшению характеристик поглощения, поэтому данный тип РПМ эффективен при маскировке от излучения РЛС, работающей на стандартной, неизменяемой моночастоте.
Нерезонансные магнитные РПМ содержат частицы феррита, распределенные в эпоксидном пластике или в покрытии. Поскольку нерезонансные магнитные РПМ рассеивают энергию высокочастотного излучения по большой поверхности, результатом является тривиальное повышение ее температуры. Иными словами уменьшение ЭПР производится за счет ухудшения ИК сигнатуры объекта. Основное преимущество нерезонансных магнитных РПМ состоит в их широкополосности — эффективности поглощения излучения в широком диапазоне частот. Напротив, эффективность резонансных РПМ ограничена узким диапазоном расчётных частот излучения /3/.
Нерезонансные объёмные РПМ обычно используются в виде относительно толстых слоев, поглощающих большую часть подводимой энергии до подхода и возможного отражения волны от металлической задней пластины. Принцип работы основан на использовании как диэлектрических, так и магнитных потерь, последнее — за счет добавления соединений феррита. В некоторых случаях используется введение графита в пенополиуретановую матрицу.
Тонкие покрытия, полученные из диэлектриков и проводников, являются узкополосными, поэтому в тех случаях, когда добавленная масса и стоимость не являются критичными, используются магнитные материалы как в резонансных РПМ, так и в нерезонансных РПМ.
Противолокационная окраска рассеивает электромагнитное излучение за счет электропроводности и ослабляет интенсивность отражения от «электромагнитных горячих точек» на поверхности самолета. Однако, высокая электропроводность обшивки самолета приводит к возникновению вторичного излучения, источником которого могут быть головки заклепок, острые кромки и выступающие антенны.
Первое сообщение о радиопоглощающих покрытиях (специальная краска, включающая ферриты) для самолета В-2 относится к 1982г.
Ведутся работы над исследованием радиопоглощающих полимеров с ретиловыми основаниями Шиффа - покрытия, которые способны снизить на 80% ЭПР самолета, оказались хорошими поглотителями радиоволн. Возможна разработка серии абсорбентов радиолокационных волн, каждый из которых рассчитан на поглощение волн узкого диапазона. Комбинация абсорбентов может обеспечивать поглощение в широком спектре радиолокационного излучения. Серьезные трудности в использовании оснований Шиффа заключаются в том, что они не растворяются и не поддаются диспергированию в плотном полиуретане, используемом для покрытия ЛА.
Отмечается, что при использовании оснований Шиффа одинаковая по сравнению с ферритами радиолокационная заметность достигается при меньшей массе материала в 10 раз /1/. Исследования показали, что при облучении самолета температура обшивки с покрытием из полимеров практически не увеличивается.
Наносимые на различные элементы самолета радиопоглощающие покрытия отличаются от тех, которые применялись 10-15 лет тому назад. Современные покрытия имеют переменную по профилю толщину, сложную структуру с меняющимися значениями диэлектрической и магнитной проницаемостей как по толщине (нормально к поверхности), так и вдоль поверхности обшивки.
Создание веществ с произвольным законом поведения магнитной проницаемости связано с некоторыми дополнительными ограничениями.
Оптимизация ЭПР какого-либо фрагмента самолета изменением диэлектрической и магнитной проницаемости вещества, распределением его толщины по поверхности фрагмента требует разработки технологий, которые в дальнейшем воспроизведут требуемые параметры.
Для нанесения покрытий используются совершенно разные технологии. Большая часть поверхности самолета покрывается методом лакокрасочных технологий. Несмотря на кажущуюся простоту, эта технология наиболее ответственная, поскольку от ее качества зависит вклад в ЭПР одного из основных рассеивающих элементов самолета - его воздухозаборника, кроме того, нарушение адгезии покрытия в воздухозаборнике может привести к аварии самолета. Многослойные радиопоглощающие покрытия напыляются с помощью как роботизированных систем в серийном производстве самолетов, так и ручным способом при создании экспериментальных образцов техники.
Для упрощения технологии нанесения покрытий предлагается не изменять его толщину, а дифференцированно распределять радиопоглощающие свойства по объему. Для этого в ряде случаев улучшить радиопоглощающие покрытия, можно используя ферромагнитные проводящие частицы очень малого размера, неравномерно распределенные как по площади, так и по глубине залегания. Распределение частиц предлагается производить после нанесения покрытия, до момента его затвердевания воздействуя на частицы силами электростатического поля. В электрическом поле на диэлектрики и проводники действуют силы. Их называют пондеромоторпыми силами, т. е. силами, действующими на весомые тела. Первопричиной возникновения пондеромоторных сил являются электрические заряды, сообщаемые телам. Однако сообщение зарядов телам осложняется появлением поляризационных зарядов и упругих деформаций в диэлектриках и проводниках. Пондеромоторные силы, действуют на границе раздела двух диэлектриков с разными диэлектрическими проницаемостями. Управление движением ферромагнитных проводящих частиц очень малого размера возможно изменением конфигурации электростатического поля. Максвеловские силы обусловленные воздействием электрополя на жидкий диэлектрик на границе раздела двух диэлектриков, что приводит жидкость в движение вдоль электродов, в результате чего в межэлектродной зоне возникает система вихрей /2/. Вихри с максимальной частотой вращения возникают у торцов фокусирующих элементов. Частицы, попадающие в подобную систему, также принимают участие в циркуляции и, как правило, имеют тенденцию сохранять свое положение в том вихре, в который они попадают при входе в систему. Это свойство обеспечивает создание работоспособной конструкции, позволяющей транспортировать частицы вдоль фокусирующей системы вплоть до выхода из нее.
Таким образом воздействуя электростатическим полем определенной конфигурации на покрытия с ферромагнитными частицами можно изменять его радиопоглощающие свойства в нужных направлениях.
Литература
1. Иванов Е.Г. Новые авиационные материалы и технологии.// М.: ВАТУ – 2008. - 97 с.
2. Леонов В.В. Исследование явления электрического ветра и применение его для перекачивания жидких диэлектриков.// Вестник Приазовского государственного технического университета – 2011. – Вып. №1/2011(13)
3. http//vpk.name/blog/fundamentalnye _i_prikladnye_ problemy _ stelstehnologii
ВУНЦ ВВС «ВВА»
УДК 574
О.Н. Болдырева
ВОПРОСЫ ИНТЕГРАЛЬНОЙ ОЦЕНКИ ЭКОЛОГИЧЕСКИХ АСПЕКТОВ И МОНИТОРИНГА ПРОМЫШЛЕННЫХ ОБЪЕКТОВ
Рассматриваются проблемы, возникающие при техническом оснащении службы экологического контроля выбор соответствующих методов и средств контроля
В настоящее время создано большое количество специальной и универсальной контрольно-измерительной и аналитической техники, технологий проведения анализа, контроля, оценки, обработки информации. В этом плане задача технико-технологического экологического контроля сводится к выбору оптимального комплекта технических средств контроля из числа серийно выпускаемых на основе нормативной базы экологического контроля.
Результаты экологического контроля составляют информационную базу экологического контроля, что позволяет использовать ЭВМ для сбора, хранения, обработки и анализа информации. Информационное обеспечение является основой для управления экологической безопасностью производств. Информативность контроля во многом зависит от уровня технических средств (оснащенности службы), поэтому при комплектовании парка приборов необходимо руководствоваться комплексом нормативов контроля объемами, периодичностью, требуемой точностью и достоверностью, полнотой. Обязательным условием обеспечения требуемой информативности – это использование ЭВМ и средств контроля на их основе.
Задача производственного экологического контроля состоит в определении количества и состава выбросов в заданный интервал времени (в течение года). Разные методы экологического контроля равноправны по критериям информативности, точности и достоверности, так как специальные исследования по сравнительной эффективности тех или иных методов контроля одного и того же параметра не проводились.
В общем случае все методы определения количества и состава содержания вредных веществ в воде и воздухе можно рассматривать как химические, физические, биологические и комплексные методы анализа веществ.
Выбор соответствующих методов и средств контроля обусловлен возможностью индикации тех или иных соединений, признанных по действующим нормам вредными для биоты. Прогноз содержания в выбросах или стоках каких-либо вредных веществ также влияет на выбор методов и технических средств экологического контроля.
Обычно состав загрязнителей воздуха, воды, почв достаточно точно прогнозируется, поэтому задача экологического контроля сводится к количественному определению концентраций известных загрязнений.
Организации экологического контроля какой-либо определенной территории с находящимися на ней источниками загрязнения должны предшествовать научно-исследовательские работы, необходимые для исследования и прогнозирования возможных загрязнений по составу и объемным концентрациям. Результаты исследований служат основанием для укомплектования экологических служб техническими средствами измерений и анализа состава загрязнителей.
Экологический контроль производств должен быть тесно связан с экологическим мониторингом (его нормативно-правовая база пока не стандартизирована и часто трактуется весьма произвольно). Экологический мониторинг должен давать ответы на следующие вопросы:
- каково состояние окружающей среды в рассматриваемый отрезок времени в сравнении с предшествующим техногенезу состоянием и какие изменения ожидаются в прогнозируемый отрезок времени;
- каковы причины происшедших изменений и возможных изменений и что является источником этих изменений;
- какие воздействия локальную природную среду, определяемые исходя из оценок функции «полезности-вредности», являются нежелательными или недопустимыми;
- какой уровень техногенных воздействий является недопустимым или критическим, после которого восстановление экологического баланса невозможно.
Организация экологического мониторинга дело будущего, так как требует значительных затрат. Но реализация экологического мониторинга позволит решить задачу регулирования качества природной среды, управление протекающими в ней техногенными и естественными процессами, а главное, реализуется планомерное повышение качества окружающей среды, социальной сферы, обеспечение максимальной экологической безопасности общества.
Поэтому необходимо выбрать стратегию регулирования и управления экологической безопасностью, экономически выгодную при самых строгих экологических нормах. Для этого необходимо решить ряд вопросов:
- к какому уровню качества природной среды следует стремиться с учетом экологической и экономической точек зрения;
- проведение каких мероприятий необходимо для снижения или полной компенсации вредных технологических процессов, снижения или полной ликвидации экологического ущерба;
- каков долговременный экологический прогноз;
- каким образом определять приоритетность действий при столкновении экономических и экологических интересов.
Комплексный экологический мониторинг повышает требования к соблюдению экологических правил и норм, позволяет реализовать систему превентивных мер и снижения экологического риска на основе аналитического прогноза фактической безопасности конкретной производственно-технической системы, что представлено на рисунке 1.
Экологический контроль должен быть многосторонним, т.е. не исключать ни одной сферы деятельности человека, так или иначе влияющей на изменение состояния окружающей среды. Экологический контроль является неотъемлемым звеном в системе промышленного производства, строительства или иного вида трудовой деятельности.
Литература
1. Бронштейн Н.М., Литвин В.А., Русин И.И. Экологизация экономики. Методы рационального управления. – М.: Прогресс, 1994.-164с.
2. Мамедов Н.М. Экология и техника (проблемы оптимальной ориентации технического развития) – М.:Знание, 1989. – 240с.
3. Охрана природной среды: Пособие для инженера-эколога. Под ред. В.И. Седлецкого и А.Д. Хованского. – Ростов-на-Дону: Изд. СКНУ ВШ, 1992.-302с.
4. Охрана окружающей среды. Под ред. К.Г.Гормана, А.А.Гусева.-М.:Экономика, 1977.-231с.
5. Шеховцов А.А., Звонов В.И., Чижов С.Г. Влияние отраслей народного хозяйства на состояние окружающей среды. – М.: Изд. Центр «Метеорология и гидрология», 1995. -186с.
Воронежский государственный технический университет
УДК 541.13
А.В. Хвостов, М.Ю. Чайка, Б.А. Спиридонов
КОНСТРУКТИВНЫЕ ОСОБЕННОСТИ СУПЕРКОНДЕНСАТОРОВ
Рассмотрен принцип работы суперконденсаторов, электрохимические процессы, протекающие в них, конструктивные особенности и области применения
Известно, что запасы углеводородов истощаются, поэтому проблема развития энергосберегающих технологий становиться все более актуальной. . С появлением суперконденсаторов (ионисторов) стало возможным использовать конденсаторы в электрических цепях не только как преобразующий элемент, но и как источник напряжения. Суперконденсатор – это электрохимическое устройство, конденсатор с органическим или неорганическим электролитом, «обкладками» в котором служит двойной электрический слой (ДЭС) на границе раздела электрода и электролита. Энергия в суперконденсаторе содержится в виде статического заряда. Для сравнения на рисунке 1 показан обычный конденсатор, состоящий из двух обкладок, изготовленных из металлической фольги. Они разделенны диэлектриком малой толщины, и имеют емкость порядка от пикофарад до микрофарад. В суперконденсаторах вместо фольги используется пористый углерод высокой удельной поверхности до тысяч кв. м на грамм. Суперконденсатор может накопить гораздо больше заряда: относительно большой аппарат имеет емкость в несколько тысяч фарад, то есть на 10 порядков величины больше, чем твердотельные конденсаторы.
Рис.1. Сравнение конструктивных схем трёх конденсаторов
В суперконденсаторах энергия накапливается в процессе зарядки за счет поляризации двойных электрических слоев (ДЭС) на границах раздела анод-электролит и катод-электролит (рис. 2).

Рис. 2. Схематическое представление суперконденсатора
В жидких электролитах ДЭС имеет толщину до 1 нм, что позволяет отнести сам принцип функционирования суперконденсатора в область наноэлектрохимии. Ёмкость современных суперконденсаторов составляет 1...10 000 Ф. Они имеют ультратонкий ДЭС (d ~ 1 нм) и гигантские площади S распределённых в объёме прибора дисперсных электродов. В качестве электродных материалов в СК используются пористые вещества с внутренней поверхностью до 1000...3000 м2/г. Ёмкость СК может быть оценена по формуле плоского конденсатора
C = = ε0εrS/d,
где ε0 = 8,85х10–12 Ф/м, εr - относительная диэлектрическая проницаемость ДЭС. Толщина ДЭС d зависит от концентрации ионов в электролите и размера ионов и для концентрированных жидких электролитов составляет 0,5...1 нм. Поэтому на гладких электродах поверхностная плотность ёмкости превышает 10–5 Ф/см2, а напряжённость электрического поля в ДЭС может быть больше 107 В/см.
Модельные представления о строении двойного слоя на границе электрод/раствор развивались в течение длительного времени. Первая работа относится к 1853 г., когда Г. Гельмгольц для описания границы между электродом и раствором предложил модель плоского конденсатора. Согласно теории Гельмгольца, к слою зарядов на металле жестко притянуты ионы противоположного знака, так что двойной слой представляет собой своеобразный плоский конденсатор с очень малым расстоянием между его обкладками (порядка диаметра молекулы воды). Эта теория предсказывала правильные по порядку величины емкости двойного слоя, объясняя форму электрокапиллярных кривых, но не могла объяснить зависимости емкости и пограничного натяжения от концентрации электролита и температуры.
Позднее Ж.Гуи и независимо от него Д. Чапмен предложили теорию диффузного слоя. В теории Гуи-Чапмена ионы рассматривались как математические точки, которые находятся под действием теплового движения и одновременно притягиваются или отталкиваются заряженной поверхностью электрода. Математически эта теория построена точно так же, как возникшая позже теория Дебая-Хюккеля. Однако в теории Гуи-Чапмена рассматривалось влияние электрического поля только вдоль одной координаты (перпендикулярной поверхности электрода). Это обстоятельство упрощало задачу и позволяло получить точное решение уравнения Пуассона-Больцмана:
![]() ,
,
где
![]() -
потенциал в пределах двойного
электрического слоя на расстоянии
х
от
поверхности металла.
-
потенциал в пределах двойного
электрического слоя на расстоянии
х
от
поверхности металла.
Теория
Гуи-Чапмена качественно объясняла
зависимость емкости и пограничного
натяжения от концентрации электролита
и температуры. Однако количественный
расчет емкости двойного слоя по этой
теории при
![]() = 1 В приводил к величинам, которые на
7-9 порядков превышали опытные значения.
Этот результат был связан с допущением
теории о том, что ионы представляют
собой частицы точечного размера, а
потому могут бесконечно близко
подходить к поверхности электрода. О.
Штерн учел собственные размеры ионов,
создав теорию, до некоторой степени
аналогичную второму приближению
теории Дебая-Хюккеля. Одновременно в
теории Штерна были учтены силы
неэлектростатического взаимодействия
ионов с металлом, что позволило
интерпретировать явления, связанные
со специфической адсорбцией ионов.
= 1 В приводил к величинам, которые на
7-9 порядков превышали опытные значения.
Этот результат был связан с допущением
теории о том, что ионы представляют
собой частицы точечного размера, а
потому могут бесконечно близко
подходить к поверхности электрода. О.
Штерн учел собственные размеры ионов,
создав теорию, до некоторой степени
аналогичную второму приближению
теории Дебая-Хюккеля. Одновременно в
теории Штерна были учтены силы
неэлектростатического взаимодействия
ионов с металлом, что позволило
интерпретировать явления, связанные
со специфической адсорбцией ионов.
Предполагалось, что двойной электрический слой состоит из двух частей: плотного и диффузного, которые разделены плоскостью, получившей название плоскости Гельмгольца. Толщина плотного слоя равна радиусу гидратированных ионов (0,3-0,4 нм), а его диэлектрическая постоянная значительно ниже диэлектрической постоянной в объеме раствора. Это обусловлено довольно жесткой ориентацией диполей растворителя в плотном слое как под действием электрического поля электрода, так и в результате их взаимодействия с металлом. В диффузном слое диэлектрическая постоянная принималась равной диэлектрической постоянной растворителя в объеме раствора. Толщина диффузного слоя теоретически бесконечна, но практически вводилась некоторая эффективная толщина, аналогичная радиусу ионной атмосферы в теории Дебая — Хюккеля.
В теории Штерна не учитывалось различие минимального расстояния, до которого могут приближаться к поверхности электрода электрические центры поверхностно-неактивных и специфически адсорбирующихся ионов. В действительности, однако, ионы, которые специфически адсорбированы, частично дегидратированы со стороны металла, а потому они входят внутрь плотного слоя, и их электрические центры располагаются ближе к поверхности электрода, чем такие же центры полностью гидратированных поверхностно-неактивных ионов. В результате вместо одной плоскости Гельмгольца необходимо было ввести две плоскости: внутреннюю и внешнюю. На внутренней плоскости Гельмгольца локализуются центры специфически адсорбированных ионов, а внешняя плоскость Гельмгольца представляет собой границу максимального приближения к поверхности электрических центров всех ионов, находящихся в диффузной части двойного слоя и участвующих в тепловом движении. Детальная модель ионного двойного слоя с учетом двух плоскостей Гельмгольца была развита в работах Д. Грэма.
Кроме разности потенциалов, создаваемой зарядами металла и ионами двойного слоя, электрические свойства границы раздела электрод/раствор зависят также от находящихся на поверхности диполей растворителя. Ориентация диполей, естественно, изменяется при изменении заряда электрода. Предполагалось существование в плотной части двойного слоя двух состояний диполей воды, ориентированных либо положительным, либо отрицательным концом к поверхности электрода. Эти диполи взаимодействовали с зарядами электрода и друг с другом по законам классической электростатики. В результате при изменении заряда электрода изменялось долевое отношение диполей с противоположными ориентациями. В дальнейшем модели этого типа уточнялись с учетом значительного числа эффектов, в том числе: возможного образования ассоциатов из двух или более молекул растворителя; наличия диполей, ориентированных параллельно или наклонно к поверхности; существования неэлектростатических взаимодействий между молекулами растворителя как в пределах ближайшего к поверхности монослоя, так и в соседних слоях.
В суперконденсаторах энергия накапливается в процессе зарядки за счет поляризации ДЭС на границах раздела «анод-электролит» и «катод-электролит». Впервые модель двойного электрического слоя в системах «электрод-электролит» создал в 1879 году Гельмгольц и показал, что ДЭС по существу является конденсатором, одна из обкладок которого — заряженная поверхность электрода, а другая — слой ионов противоположного знака в электролите (ионного проводника). Впоследствии усилиями Гуи, Штерна и Фрумкина создана классическая теория строения и свойств ДЭС в водных электролитах, и, таким образом, был заложен фундамент для создания различных электрохимических преобразователей энергии и информации в том числе и суперконденсаторов. Система «электронный проводник — ионный проводник» в определенных условиях ведет себя как конденсатор, то есть при прохождении через такую систему тока изменяется межфазная разность потенциалов φ. Если эти изменения обратимы, то система может характеризоваться емкостью, определяемой для идеального конденсатора по формуле:
С = Dφ/Dq,
где Dφ — изменение межфазной разности потенциалов, Dq — накопленный на межфазной границе заряд.
В концентрированных электролитах заряд на межфазной границе образован избыточным электронным зарядом поверхности металлического электрода и избыточным ионным зарядом со стороны электролита. Ионы электролита плотно прижаты к поверхности электрода как силами изображения, так и электростатическим притяжением электронного заряда поверхности, так что расстояние между зарядами в двойном слое по порядку величины близко к радиусу иона. В электролитах ионы, как правило, сольватированы, что несколько увеличивает их радиус. Оценка диэлектрической проницаемости в области двойного слоя в системах «электронный проводник — ионный проводник» по формуле плоского конденсатора дает величину ε = 4,5, тогда как для воды ε = 80: ε = Суд. × δ/ε0 = = 0,2 × 2 × 10–10/8,85 × 10–12 = 4,5.
Низкую величину ε объясняют тем, что молекулы воды на межфазной границе сильно поляризованы, при этом радиус гидратированных ионов принят равным 2 Å.
Если в обычных конденсаторах заряды разделены диэлектриком, то в ДЭС разделение зарядов на межфазной границе обусловлено термодинамической невозможностью или кинетической затрудненностью переноса зарядов в рабочем интервале электродных потенциалов Δφ. Сопротивление переносу R можно в этом случае выразить как R = Δφ/I утечки. В реальных системах ток утечки (I утечки) отличается от нуля из-за наличия примесей в электролите или электродах с более низким потенциалом разложения, а также из-за наличия электронной составляющей проводимости в ионных проводниках (электролитах). Электрическую прочность ДЭС можно определить по формуле Е = Δφ/δ. В различных типах ионисторов используются ДЭС с интервалом потенциалов от 0,5 до 1,5 В. Если принять Δφ = 1,0 В, а δ = 2×10–10 м, то электрическая прочность ДЭС составит:
Е = 0,5 × 1010 В/м = 5000 МВ/м = 5 ГВ/м.
Таблица 1
Величины электрической прочности диэлектриков других типов конденсаторов
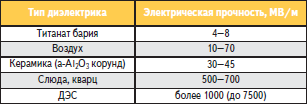
Существует три типа суперконденсаторов: 1 ) Тип 1 - с идеально поляризуемыми углеродными электродами («идеальные» суперконденсаторы). Примеры электрохимических систем:
– С / 30% водный раствор КОН / С +;
– С / 38% водный раствор Н2SO4 / C +;
– C / Органический электролит / С +.
В этом типе ионисторов на электродах в рабочем интервале напряжений не протекают электрохимические реакции, накладывающие известные ограничения на скорость зарядки и разрядки, поэтому по величине энергии и мощности, температурному диапазону и количеству циклов они ближе остальных типов к области оксидно-электролитических конденсаторов.
2) Тип 2 - суперконденсатры с идеально поляризуемым углеродным электродом и неполяризуемыми или слабо поляризуемыми катодом или анодом («гибридные» суперконденсаторы).
Примеры электрохимических систем:
– Ag / Твердый электролит RbAg4I5 / C +;
– С / 30% водный раствор КОН / NiOOH +
В этом типе на одном из электродов протекает электрохимическая реакция (как в аккумуляторах), поэтому их называют гибридными суперконденсаторами . Ёмкость ионисторов типа 2 в два раза выше, чем ионисторов типа 1, так как емкость неполяризуемого электрода замкнута сопротивлением протекающей электрохимической реакции. Эта реакция накладывает диффузионные и кинетические ограничения на скорость зарядки и разрядки суперконденсатора типа 2.
3) Тип 3 - псевдоконденсаторы — это ионисторы, на поверхности электрода которых при заряде и разряде протекают обратимые электрохимические процессы хемосорбция или интеркаляция ионов, содержащихся в электролите.: Примеры электрохимических систем
– Ni(H) / 30% водный раствор КОН / NiOOH +;
– С(Н) / 38% водный раствор Н2SO4 / PbSO4(РbO2) + .
Удельная энергия псевдоконденсаторов, в следствии протекания электрохимических реакций на обоих электродах, сравнима с энергией, накапливаемой в аккумуляторах, однако величина удельной мощности и количество циклов в режиме «зарядка-разрядка» могут быть на порядок выше того, что достигнуто в области аккумуляторов, так как диффузионные и кинетические ограничения удается минимизировать за счет увеличения площади поверхности электродов. По величине удельной энергии и мощности, температурному диапазону эксплуатации и количеству циклов псевдоконденсаторы ближе всех остальных типов конденсаторов к аккумулятрам. Необходимо отметить, что деление суперконденсаторов на три типа позволяет ориентироваться в большом многообразии этих изделий как по типу используемых электрохимических систем, так и по эксплуатационным свойствам.
Одной из основных проблем, которые препятствуют широкомасштабному внедрению суперконденсаторов в производство, является их высокая стоимость, однако в последние годы эта проблема отходит на второй план (рис. 3).
В заключении следует отметить, что создание суперконденсаторов сверхвысокой емкости предполагает различные области их применения, но в первую очередь включает системы стартерного пуска двигателей внутреннего сгорания и быстрозарядные тяговые батареи для перспективного электротранспорта. Суперконденсаторы нашли также применение в устройствах сглаживания скачков напряжения. Суперконденсаторы являются самыми оптимальными источниками питания для электротранспорта. В разработках электромобилей как российские, так и зарубежные конструкторы предусматривают использование именно суперконденсаторов. Многие аналитики предсказывают, что на ближайшие 50 лет на рынке будут доминировать гибридные автомобили с суперконденсаторными батареями сверхвысокой емкости.
Таблица 2
Преимущества и недостатки суперконденсаторов
Преимущества |
Недостатки |
Большой срок службы |
не обеспечивают достаточного накопления энергии |
Малое внутреннее сопротивление |
маленькая энергетическая плотность |
Быстрый заряд - в течение нескольких секунд |
низкое напряжение на некоторых типах ионисторов |
Работа при любом напряжении не превосходящем номинального |
для получения требуемого напряжения необходимо последовательное подключение не менее трех ионисторов |
неограниченное число циклов заряд/разряд |
высокий саморазряд |
отсутствие необходимости контроля за режимом зарядки |
|
использование простых методов заряда |
|
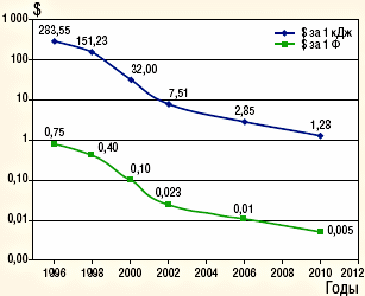
Рис. 3. Тенденции снижения стоимости 1 кДж запасаемой энергии и 1Ф емкости ЭХК к 2012 году
Литература
Меркулов В.И. Основы конденсаторостроения. Учебн. пособие. Томск. Изд.- во ТПИ. 2001. 121 с.
Меркулов В.И. Основы конденсаторостроения. Лаборат. Практикум. Изд.-во ТПИ, 2011. 84 с.
В. Кузнецов, О. Панькина, Н. Мачковская, Е. Шувалов, И. Востриков. Конденсаторы с двойным электрическим слоем (ионисторы): разработка и производство. Компоненты и технологии №6, 2005.
Воронежский государственный технический университет
УДК 630.2
О.Н. Болдырева
ОРГАНИЗАЦИЯ САНИТАРНО-ЗАЩИТНЫХ ЗОН ПРЕДУПРЕЖДЕНИЯ ВРЕДНОГО ВОЗДЕЙСТВИЯ ХИМИЧЕСКИХ ВЕЩЕСТВ НА ЭКОЛОГИЧЕСКУЮ БЕЗОПАСНОСТЬ
По мере совершенствования технологии производств и способов очистки выбросов в атмосферу роль СЗЗ как воздухоохранного мероприятия существенно уменьшилась. Однако технологические усовершенствования не обеспечивают полного исключения выбросов, поэтому в настоящее время и в будущем организация СЗЗ как метод рассеивания приземных концентраций до нормативных (безопасных) значений не теряет своей актуальность
Ныне действующие Санитарно-эпидемиологические правила и нормативы (СанПиН 2.2.1/2.1.1.1200-03 «Санитарно-защитные зоны и санитарная классификация предприятий, сооружений и иных обьектов», далее СанПиН) [1] «устанавливают гигиенические требования к размеру санитарно-защитных зон в зависимости от санитарной классификации предприятий, сооружений и иных обьектов, требования к их организации и благоустройству, основания к пересмотру этих размеров».
«Предприятие» и «производство» выступают в качестве объектов хозяйственной деятельности, производящих определенный объем продукции или услуг за определенное время. Следовательно, их класс и размер ширины СЗЗ могут устанавливаться в зависимости от этой мощности и других критериев, в том числе качественного и количественного состава выбросов, значений гигиенических нормативов.
Весьма часто «производство», да и «предприятие» фигурирует как процесс или определенный характер (вид, тип) деятельности: «производство аллюминия способом электролиза расплавленных солей аллюминия. Однако использование этого устаревшего критерия ограничивает санитарную классификацию предприятий и производств, так как по их характеру можно определить только одно значение класса и соответствующую ему ширину СЗЗ.
Чаще всего это наиболее опасный класс с наибольшей шириной СЗЗ, что не соответствует современным условиям развития рыночной экономики в Российской Федерации, когда существенно изменяются мощность, технология предприятий, очистка, качественный и количественный состав выбросов.
Определяя класс конкретного предприятия или производства только по характеру его деятельности, нельзя утверждать, что этот объект включен в санитарную классификацию.
Между тем в СанПиН отсутствует механизм определения класса опасности предприятия. Поэтому размер ширины СЗЗ устанавливается по «результатам расчетов ожидаемого загрязнения атмосферного воздуха и уровней физических воздействий». При необходимости класс предприятия следует определять по установленному размеру ширины СЗЗ.
Опасность предприятий и производств – это вероятность (риск) неблагоприятного воздействия на здоровье населения химического, физического и биологического загрязнения, уровень которого уменьшается до нормативных значений путем рассеивания за счет увеличения расстояния от его источников.
Опасность предприятий и производств как потенциальных носителей неблагоприятного воздействия на здоровье населения исторически нашла свое отражение в размерах ширины их санитарно-защитных зон и сани тарной классификации.
Санитарно-защитная зона – территория, ширина которой, измеряемая от источников химического, биологического и физического загрязнения окружающей среды до ее границы в заданном направлении, обеспечивает уменьшение указанного загрязнения до значений гигиенических нормативов и (или) величин приемлимого риска для здоровья населения.
Класс предприятия и производства – количественная характеристика опасности предприятия или производства, имеющая порядковое числовое значение и соответствующую ему ширину санитарно-защитной зоны: первый класс – ширина 1000м; второй класс – 500м; третий – 300м; четвертый – 100м; пятый – 50м [1,2].
Порядковые номера классов предприятий и производств обратно пропорциональны размеру ширины СЗЗ, т.е, в традиционном обозначении с увеличением порядкового номера класса опасность предприятий уменьшается, что следует учитывать при поиске количественной связи опасности предприятий с их классами.
Установление такой связи устраняет существенный недостаток санитарной классификации: дискретный характер принятых значений классов и соответствующих им размеров ширины СЗЗ, являющихся величинами непрерывными [3]. Это ведет к тому, что в случае незначительных изменений опасности предприятия вблизи границ диапазона класса, например по критерию мощности или другому критерию, происходит неоправданно резкое формальное изменение характеристики класса и размера ширины СЗЗ.
В целях устранения дискретности значений классов, размеров ширины СЗЗ и относительной опасности предприятий необходимо установить зависимость размеров ширины СЗЗ как отражения опасности предприятий от их классов.
Для обеспечения соответствия традиционного обозначения классов степени опасности предприятий можно предложить формулу, позволяющую получить по традиционному порядковому числу класса (zj) его скорректированное числовое значение (xj), соответствующее опасности класса: (xj = 6 – zj).
Логический и графический анализ показывает, что зависимость минимальной безопасной для здоровья населения СЗЗ предприятия от его класса (скорректированного числового значения) может быть представлена уравнением: lgyj = 1,3979 + 0, 3242xj. (1)
На основе сопоставления мощности и соответствующих классов указанных предприятий установлено, что скорректированный класс опасности конкретного предприятия (xj) как непрерывная величина равен отношению суммы его мощности (М) и коэффициента пропорциональности классов (К = 1999 т/год) к числу, определяющему диапазон градации класса
В соответствии с формулой (1) получена формула (2), позволяющая определять с учетом класса предприятия (xj) его опасность как относительную вероятность (yj) возникновения неблагоприятных эффектов у населения, проживающего в районе размещения предприятия, когда меры по созданию санитарно-защитной зоны не предпринимаются: lg yj = 0,3242xj (2)
Согласно формуле (2), относительная вероятность неблагоприятных эффектов возрастает по отношению к ее фоновому уровню, равному 1: у предприятий 5 класса в традиционном обозначении – в 2,11 раза; 4 класса – в 4,3 раза; 3 класса – в 9,4 раза; 2 класса – в 19,8 раза; и 1 класса – в 41,8 раза, что удовлетворительно совпадает с оценкой опасности предприятий разных классов как отношения нормативных размеров ширины их СЗЗ.
Вместе с тем следует отметить, что оценка опасности предприятий чрезвычайно сложна, так как требует всестороннего анализа обусловленого ими загрязнения во времени и в пространстве с качественной и количественной характеристиками его компонентов, особенностей их биологического действия, режима и длительности воздействия на население, величин гигиенических нормативов, сообенностей источников загрязнения и селитебных зон, их размещения на территории, а так же метеорологических условий и многого другого.
Например опасность предприятий может изменяться на 7-9 порядков только в зависимости от величины гигиенических норматтивов загрязняющих веществ при равенстве всех прочих условий.
Решению этих сложнейших задач может способствовать дальнейшее развитие технологий моделирования загрязнения окружающей среды в целом и атмосферного воздуха в частности.
Как свидетельствует практика, указанные требования по многим объективным причинам не выполнимы или выполняются формально, не обеспечивая хотя бы относительно достоверных результатов [3-6].
Для контрольных же целей наиболее важна автоматическая регистрация содержания загрязняющих веществ в выбросах. По всем условиям такой контроль обеспечивает получение объективных данных и с учетом моделей рассчета рассеивания выбросов позволяет характеризовать загрязнение во времени и пространстве, что при ограниченных финансовых и трудовых ресурсов с помощью натурных исследования загрязнения атмосферы получить невозможно.
Установление размера ширины СЗЗ в зависимости от источников осуществляется «в случае наличия только высоких нагретых выбросов». Однако установление размера СЗЗ от границы промплощадки не столь однозначно, ибо при требовании установления СЗЗ от границы промплощадки не исключается возможность установления размера СЗЗ от источников.
В этих условиях установление СЗЗ от источника выбросов получает наибольшую обоснованность, хотя в этом случае можно устанавливать СЗЗ и от границы промплощадки.
В любом случае при установлении ширины СЗЗ от границы промплощадки расстояние от источника (источников) до границы СЗЗ и, следовательно, до жилой застройки будет больше, чем при установлении ширины СЗЗ от источников выбросов.
Так, известная «Методика рассчета концентраций в атмосферном воздухе вредных веществ, содержащихся в выбросах предприятий» (ОНД-86) свидетельствует, что кроме самого источника выбросов никакой другой точки отстчета для получения значения максимальной концентрации на определенном расстоянии быть не может [7].
Имеются формулы позволяющие по заданному уровню максимальной приземной концентрации при прочих фиксированных параметрах выброса определять мощность и минимальную высоту источника выбросов или установить нормативы предельно допустимых выбросов, или размер санитарно-защитной зоны предприятия.
В настоящее время указанная система формул и построение изолиний полей концентраций с помощью компьютерных геоинформационных систем позволяют решать любые задачи по установлению расстояний, на которых уровни концентраций достигают любых заданных значений, в том числе равных значениям гигиенических нормативов любого периода осреднения по времени.
Установление минимальных размеров СЗЗ отражает пассивность градостроительных мер для защиты здоровья населения.
Поэтому вместо установления минимальных размеров ширины СЗЗ следует использовать принцип их оптимальной минимизации [3,10], когда внедрение современных технологий и методов очистки выбросов становится требованием, а не пожеланием, как это звучит в п. 2.3 СанПиН: «Обязательным условием современного промышленного проектирования является внедрение передовых ресурсосберегающих, безотходных и малоотходных технологических решений, позволяющих максимально сократить или избежать поступлений вредных химических или биологических компонентов выбросов в атмосферу, почву и водоемы, предотвратить или снизить воздействие физических факторов до гигиенических нормативов и ниже».
При оптимальной минимизации ширины СЗЗ в условиях рыночной экономики предприятия и производства любой формы собственности становятся заинтересованными во внедрении технологий, сокращающих объемы выбросов.
Вызывает недоумение противоречивость требования пункта 2.23 СанПиН: «не допускается размещать» предприятия по производству лекарственных веществ, лекарственных средств и (или) лекарственных форм, складов сырья и полупродуктов для фармацевтических предприятий там, где концентрация выбросов предприятий других отраслей промышленности будут выше 0,1 ПДК. Во-первых, в соответствии с этим требованием качество воздуха в районе размещения указанных объектов должно быть в 10 раз лучше качества атмосферного воздуха жилых зон и в 8 раз – курортных зон. Во-вторых, указанные объекты, согласно СанПиН, сами могут относиться к предприятиям разных классов, в том числе 1-го класса («производство синтетических химико-фармацевтических и лекарственных препаратов») и быть источниками разнообразных загрязняющих веществ.
Вообще проблема использования территорий СЗЗ и их организации нуждается в специальной научной разработке, так как охрана здоровья населения – это не только запретительные меры, порой основанные на использовании двойных стандартов. Так, согласно п. 2.25 размещение спортивных сооружений, парков, образовательных и детских учреждений, лечебно-профилактических и оздоровительных учреждений общего пользования на территории санитарно-защитной зоны не допускается. Однако согласно п. 2.26 в границах санитарно-защитной зоны допускается размещать: учебные заведения, поликлиники, спортивно-оздоровительные сооружения для работников предприятия. Принципиально для таких работников нужны условия, способствующие максимальному снижению дозы экспозиции. Слудовательно вопрос использования территорий СЗЗ должен осуществляться с большой гибкостью, с учетом конкретных условий, ибо известно, что снижение концентраций перед границей СЗЗ происходит плавно, а не ступенькой.
Литература
СанПиН 2.2.1/2.1.1.1200-03 «Санитарно-защитные зоны и санитарная классификация предприятий, сооружений и иных объектов». – М., 2003.
СН 245-71 «Санитарные нормы проектирования промышленных предприятий». – М., 1972
Принципы установления размеров санитарно-защитных зон предприятий, не включенных в санитарную классификацию // Гигиеническая наука и практика на рубеже XXI века: Мат. IX Всерос. Съезда гигиенистов и санитарных врачей. - М., 2001. Т. 1. С. 560-563.
Классификация производств и предприятий с установлением размера санитарно-защитных зон как градостроительная форма защиты населения от воздействия вредных производственных факторов / М.А. Пинигин и др. // Итоги и перспективы научных исследований по проблеме экологии человека и гигиены окружающей среды. – М., 2002. С.107-124.
Критерии и принципы установления размеров санитарно-защитных зон промышленных предприятий / М.А. Пинигин и др. // Современные проблемы профилактической медицины, среды обитания и здоровья населения промышленных регионов России. – Екатеринбург, 2004. С.97-102.
Санитарная классификация предприятий и проблема установления санитарно-защитных зон / М.А. Пинигин и др. // Гигиена и санитария. 2003. № 6. С. 22-24.
ОНД-86 «Методика расчета концентраций в атмосферном воздухе вредных веществ, содержащихся в выбросах предприятий». – Л.: Госкомгидромет, 1987.
СанПиН 2.2.1/2.1.1.1200-03 «Санитарно-защитные зоны и санитарная классификация предприятий, сооружений и иных объектов». – М., 2003.
Воронежский государственный технический университет
УДК 621. 367. 502.7
И. М. Винокурова, Д.Н. Бурдунюк
ИЗУЧЕНИЕ ДИФФУЗИОННОЙ КИНЕТИКИ И КОНЦЕНТРАЦИОННОЙ ПОЛЯРИЗАЦИИ ПРИ РАЗЛИЧНЫХ режимах ОБРАБОТКИ
В материалах статьи приводятся данные по определению процессов анодной обработки с учетом диффузии и концентрационной поляризации
Электродные процессы электрохимической обработки металлов обязательно включают в себя, помимо электрохимической реакции, стадии массопереноса, осуществляемые диффузией или конвекцией: отвод продукта анодного процесса (ионов металла) от места реакции - поверхности металла, перенос, частиц деполяризатора катодного процесса к поверхности металла и отвод продуктов катодной деполяризационной реакции от места реакции - поверхности металла в глубь раствора и т. п. В электрохимических процессах довольно часты случаи диффузионного или диффузионно-кинетического контроля, т. е. значительной заторможенности стадий массопереноса. В связи с этим диффузионная кинетика представляет теоретический и практический интерес.
Согласно
теории Нернста, к поверхности твёрдого
тела прилегает тонкий слой
жидкости
толщиной
![]() ,
в котором происходит диффузия
растворяющегося вещества. За пределами
этого слоя движение жидкости, увлекающей
растворенное вещество, приводит к
поддержанию постоянства концентрации
во всем остальном объеме раствора.
Толщина
и получила название толщины диффузионного
слоя Нернста. Она зависит только от
скорости перемещения диффундирующего
вещества
,
в котором происходит диффузия
растворяющегося вещества. За пределами
этого слоя движение жидкости, увлекающей
растворенное вещество, приводит к
поддержанию постоянства концентрации
во всем остальном объеме раствора.
Толщина
и получила название толщины диффузионного
слоя Нернста. Она зависит только от
скорости перемещения диффундирующего
вещества
![]() (1)
(1)
где
v
-скорость
движения жидкости; п=1/2![]() 1.
1.
Распределение концентраций в диффузионном слое имеет линейный характер (рис. 1).
В
обычных условиях перемешивания
=![]() см, что соответствует десяткам тысяч
молекулярных слоев. Такой слой не может
удерживаться молекулярными силами.
Кроме того, прямые опыты показали, что
на расстояниях порядка
см, что соответствует десяткам тысяч
молекулярных слоев. Такой слой не может
удерживаться молекулярными силами.
Кроме того, прямые опыты показали, что
на расстояниях порядка
![]() см от твердой стенки наблюдается движение
жидкости, а следовательно, линейный
закон распределения концентрации теряет
свое обоснование. Теория Нернста не
позволяет оценить значение потока т
теоретически,
так как толщина
в ней не вычисляется, поэтому теория
является только качественной, а не
количественной.
см от твердой стенки наблюдается движение
жидкости, а следовательно, линейный
закон распределения концентрации теряет
свое обоснование. Теория Нернста не
позволяет оценить значение потока т
теоретически,
так как толщина
в ней не вычисляется, поэтому теория
является только качественной, а не
количественной.
Скорость диффузии в приэлектродном слое в направлении х, нормальном к поверхности электрода, дается первым законом Фика:
![]() (2)
(2)
г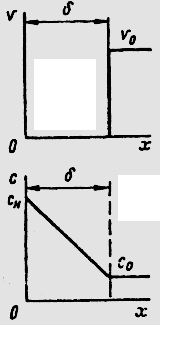 де
-
де
-![]() - градиент концентрации; S=
SMe
— сечение потока диффузии, равное
площади электрода.
- градиент концентрации; S=
SMe
— сечение потока диффузии, равное
площади электрода.
Рис. 1. Зависимость скорости движения жидкости v и концентрации раствора с от расстояния (по Нернсту)
Учитывая, что < 0, и полагая S = 1 см2, можно получить
удельную скорость диффузии:
![]() (3)
(3)
Если принять, по Нернсту, линейное изменение концентрации, можно уравнение (3) записать следующим образом:
![]() (4)
(4)
При толщине диффузионного слоя б (расстояние, на котором с претерпевает линейное изменение от с0 до с - рис. 2) и разности концентраций си — с, предполагая молекулярную диффузию в слое толщиной б и конвективный перенос в остальном объеме электролита, получим следующее уравнение:
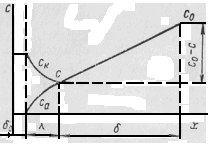
![]() (5)
(5)
Рис. 2. Распределение эквивалентной концентрации катионов (ск), анионов (cа) и электролита в целом с вблизи поверхности электрода
Движение жидкости относительно электрода стабилизирует толщину диффузионного слоя δ и делает ее меньше, что соответствует конвективной диффузии, т. е. диффузии в движущейся жидкости. Увеличение скорости перемещения жидкости приводит к ускорению диффузии.
Из гидродинамики известно, что скорость жидкости, обтекающей твердую поверхность, только в непосредственной близости от нее равна нулю, а далее постепенно возрастает и достигает величины, свойственной потоку жидкости (рис. 3). Этот слой жидкости с постепенно нарастающей от нуля до υ0 скоростью движения, толщиной П, называют граничным слоем Прандтля.
Из
гидродинамической теории следует, что
толщина граничного слоя Прандтля зависит
от скорости движения жидкости относительно
твердого тела υ0
и
кинематической вязкости жидкости ν
(![]() ).
Кроме того, эта толщина зависит от
расстояния рассматриваемой точки х
от
точки набегания струи на плоский
электрод. По мере увеличения этого
расстояния х
толщина П
возрастает,
согласно уравнению
).
Кроме того, эта толщина зависит от
расстояния рассматриваемой точки х
от
точки набегания струи на плоский
электрод. По мере увеличения этого
расстояния х
толщина П
возрастает,
согласно уравнению
![]() (6)
(6)
до какого-то расстояния х, после чего она стабилизируется, т. е. П =const (рис.4).
Перенос вещества в движущейся жидкости обусловлен двумя разными механизмами:
при наличии разности концентраций и жидкости возникает молекулярная диффузия, т. е. самопроизвольное выравнивание концентрации;
частицы растворенного в жидкости вещества увлекаются последней в процессе ее движения (конвекции) и переносятся вместе с ней.

Рис. 3. Распределение скорости движения жидкости вблизи поверхности твердого тела |
Рис. 4. Изменение толщины граничного слоя Прандтля вдоль поверхности пластинки, обтекаемой жидкостью |
На рис. 5 показаны конвекционные потоки, возникающие в называемой обычно неподвижной (неперемешиваемой) теплой воде вследствие охлаждения последней возле стенок сосуда, что делает ее более тяжелой и заставляет опускаться вниз, а па ее место поступает более теплая вода из центральном части сосуда. Это самоперемешивание неподвижной жидкости можно наблюдать, если в ней имеются пылинки или другие мелкие частицы (например, волоски ваты) при пропускании через сосуд яркого света, например солнечного. При приближении температуры общей массы поды к комнатной эти конвекционные потоки ослабевают, по поддерживаются за счет охлаждения воды ее испарением с поверхности (скрытая частота испарения воды Qисп=539 кал/г). Если в сосуде не вода, а раствор, то вследствие испарения воды с поверхности происходит дополнительное (помимо охлаждения) утяжеление его поверхностного слоя в результате изменения концентрации, сопровождающееся опусканием вниз. Все это снижает воспроизводимость результатов опытов по диффузии в неподвижных растворах из-за непостоянства толщины граничного слоя Прандтля жидкости у поверхности твердого тела (электрода) и толщины части ее, называемой диффузионным слоем.
Более воспроизводимые результаты получаются для перемешиваемой с определенной контролируемой скоростью жидкости и, в частности, для вращающегося дискового электрода, для которого и была в первую очередь сформулирована теория конвективной диффузии.

Рис. 5. Конвекционные потоки в неподвижной теплой воде |
Рис. 6. Распределение концентрации и касательная слагающая скорости движения жидкости у поверхности твердого тела |
В объеме жидкого электролита с постоянной концентрацией перенос вещества осуществляется конвекцией, т. е. движением жидкости. При наличии градиента концентрации в слое жидкости становится возможным перенос молекулярной диффузией.
Таким образом, в граничном слое Прандтля при наличии в нем градиента концентрации массоперенос осуществляется двумя разными параллельно протекающими путями. Суммарная скорость процесса массопереноса определяется скоростью протекания каждого элементарного процесса переноса. Если, однако, торможение одного из этих параллельных процессов значительно меньше торможения другого, то суммарная скорость массопереноса определяется в основном скоростью этого наименее заторможенного, т. е. быстрого, процесса переноса. Скорость конвективного массопереноса в граничном слое Прандтля снижается по мере уменьшения скорости движения υ в нем жидкости (см. рис. 8.8) и его роль в определении суммарной скорости массопереноса тоже уменьшается, а роль молекулярной диффузии возрастает. Начиная с какого-то расстояния от твердой поверхности δ молекулярный перенос вещества становится преобладающим по сравнению с конвективным переносом, который преобладает в части слоя Прандтля (П -δ).
Таким образом, диффузионный слой, являющийся частью слоя Прандтля, не неподвижный, а движущийся с уменьшающейся до нуля скоростью, но массоперенос в нем осуществляется в основном за счет молекулярной диффузии, которая значительно превосходит массоперенос за счет конвекции (движения) жидкости, т. е. это область наиболее резкого изменения концентрации вещества.
Теория конвективной диффузии учитывает молекулярную диффузию, идущую как поперек слоя, так и в тангенциальном направлении, вдоль него, и дает для толщины диффузионного слоя следующее уравнение:
![]()
![]() ,
(7)
,
(7)
где Рr=ν/kД — безразмерное число Прандтля, которое у воды и сходных с ней жидкостей равно примерно 103, ав вязких жидкостях достигает значения порядка 106 и более.
Так как толщина диффузионного слоя δ зависит от расстояния х до переднего края обтекаемой потоком жидкости пластинки, различные точки этой пластинки не равнодоступны в диффузионном отношении (рис. 7) тангенциальный перенос вещества движущейся жидкостью весьма существенно увеличивает эффективность диффузионного потока, который у переднего края пластинки больше, чем у заднего.
Рис. 7. Диффузионный поток m на различных расстояниях х от края пластинки
При постоянном режиме перемешивания, диффузии определенных частиц и постоянной температуре толщина диффузионного слоя δ - const; при этом изменение концентрации в диффузионном слое, как указывалось выше, почти линейно.
Замедленность диффузионной стадии электрохимического процесса приводит к возникновению концентрационной поляризации, значение которой для неконцентрированных растворов можно представить уравнением, соответствующим эдс концентрационного элемента:
ΔVконц=![]() ,
(8)
,
(8)
где с - концентрация диффундирующего вещества вблизи поверхности корродирующего металла; с0 - то же, в объеме раствора.
Для смешанного диффузионно-кинетического и чисто диффузионного (с≈0) контролей можно написать следующие уравнения:
i = k (с0 - с); (9)
iд = kc0, (10)
где iд - предельная диффузионная плотность тока. Находим из этих уравнений значения сo и с:
с0=iД/k;
![]()
и, подставляя их в уравнение (8), получаем для неконцентрированных растворов уравнение
![]() .
(11)
.
(11)
График этой зависимости приведен на рис. 8.
Рис. 8. Зависимость концентрационной поляризации от плотности тока
Концентрационная
поляризация всегда имеет место при
электрохимических электродных процессах,
увеличивая значение поляризации данного
процесса на меньшую или большую величину
(![]() ),
а часто (при высоких, близких к iд
плотностях тока) определяет суммарную
скорость процесса (диффузионный
контроль процесса).
),
а часто (при высоких, близких к iд
плотностях тока) определяет суммарную
скорость процесса (диффузионный
контроль процесса).
Если заторможенности электродной реакции и диффузии соизмеримы, то суммарная скорость электрохимического процесса будет зависеть от обеих этих стадий (смешанный диффузионно-кинетический контроль), т. е. поляризация процесса будет смешанной.
Литература
1. Жук Н.П. Курс теории коррозии и защиты металлов/ Н.П. Жук. М.: Металлургия, 1976. 474 с.
2. Фрумкин А.Н. Избранные труды: Электродные процессы/ А.Н. Фрумкин. М.: Наука, 1987. 336 c.
3. Скорчеллетти В.В. Теоретическая электрохимия/ В.В. Скорчеллетти. Л.: Госхимиздат, 1963. 608 с.
Воронежский государственный технический университет
УДК 628.5
О.Н. Болдырева, В.В. Пусовская
ИНТЕГРАЛЬНЫЙ МЕТОД АНАЛИЗА ЭКОЛОГИЧЕСКИ ЗНАЧИМЫХ АСПЕКТОВ ОПАСНЫХ ПРОИЗВОДСТВЕННЫХ ОБЪЕКТОВ
Рассматривается интегральный метод анализа для количественной оценки влияния отдельных факторов, что позволяет установить значимость факторов «машина», «окружающая среда », «человек»
Часто используемая оценка условий труда средневзвешенными показателями требует приведения значений всех производственных факторов к общим единицам измерения, что сделать довольно трудно. Поэтому целесообразно использовать балльную оценку, которая позволяет оперировать любым числом факторов и их количественным результатом. Показателем, отражающим количественные признаки опасности, служит вредность производственного фактора- количественная мера, характеризующая его значимость в совокупном воздействии комплекса вредных факторов. Вредное воздействие определяется в целях установления наиболее значимых факторов и их влияния на результативные показатели. При интегральной количественной оценки влияния факторов на результативный показатель имеют значения следующие параметры: отклонение фактического значения вредного фактора относительно предельно допустимого; значимость вредного воздействия каждого фактора; величина, учитывающая усиление влияния на организм человека рассматриваемого фактора при комбинированном воздействии.
Применения интегрального метода анализа обусловлена тем, что значение фактора, выраженное через интегральный балл, может быть представлено в виде весовых характеристик, а также соблюдается положение о независимости факторов.
Интегральный метод - он дает общий подход к решению задач самого разного вида, независимо от числа элементов, входящих в модель факторной системы и формы связи между ними, выявляет производственные факторы, влияющие на результативный показатель.
Интегральный метод анализа- теоретическая основа для количественной оценки влияния отдельных факторов на изменение результативного показателя.
В частности, он позволяет установить значимость факторов подсистемы «машина», «окружающая среда», «человек» при работе с ручным пневматическим инструментом ударного действия в процессе комплексного их воздействия на результативный показатель- вибротравматизм.
Интегральный балл ∆уi, благодаря которому можно оценить условия труда рабочих виброопасных профессий, определим по формуле:
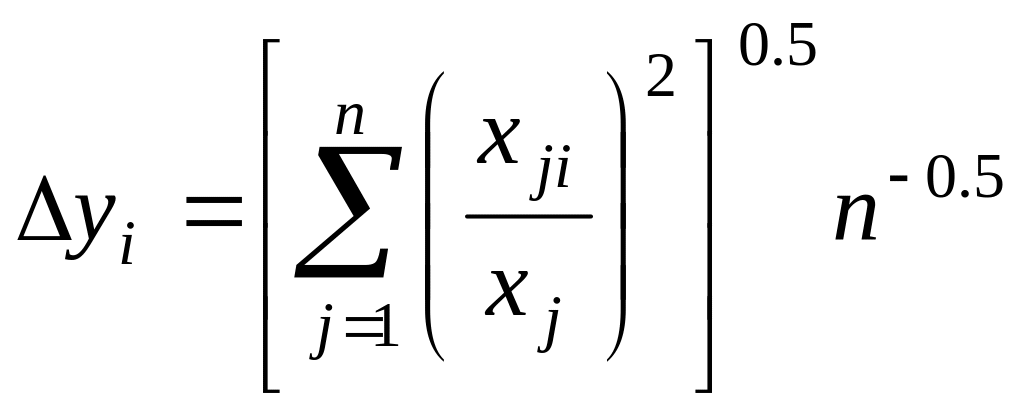 ,
(1)
,
(1)
где xji-измеренное i-е значение j-го неблагоприятного фактора производствен ной среды; xj- предельно допустимое значение j-го фактора производственной среды; n- число измерений j-го фактора.
Допустим, что при ∆yn=1 неблагоприятное воздействие факторов отсутствует, тогда при ∆yn 1 воздействие факторов следует считать значимым. Расчет интегрального балла вредности каждого фактора и комплексной нагрузки производственной среды основан на среднестатистических значениях показателей вредных факторов, формирующих условия труда.
Факторы, формирующие условия труда в подсистеме «человек», представлены в таблице. Определим интегральный балл влияния каждого фактора, т.е. его значимость в данной подсистеме, на условия труда оператора.
Для количественного определения вредности факторов введем допущение об их нормативных значениях. С учетом основных биологически значимых факторов условий труда и с использованием показателей соответствия предельно допустимым значениям определим отклонение фактического значения вредных факторов относительно нормативного (интегральный балл), т.е. значимость фактора, влияющего на условия труда в подсистеме «человек», используя формулу (1).
Факторы, формирующие условия труда, по значимости располагаются в следующей последовательности (см. таблицу): статическое напряжение мышц (∆yс=1,3) - оказывает большее влияние на возникновение вибротравматизма; условие нажатия на рукоятку (∆yу=1,21); неудобная рабочая поза (∆yп=1,16); физическая нагрузка (∆yF=1,08). Их следует считать неблагоприятными и оказывающими сопутствующее воздействие на результат в последовательности, определенной интегральным баллом.
Чтобы найти совокупное неблагоприятное воздействие факторов на условия труда, применяем формулу:
 (2)
(2)
где dj-фактическое среднее значение j-го неблагоприятного фактора; m-число определенных факторов.
Для подсистемы «человек» совокупное воздействие факторов составляет 1,46.
Фактор |
Значение фактора |
Интег-ральный балл ∆yj
|
||||||||||||
фактическое |
Предельно доступное |
|||||||||||||
Xj1 |
Xj2 |
Xj3 |
Xj4 |
Xj5 |
Xj |
|||||||||
Подсистема «человек» |
||||||||||||||
Статическое напряжение мышц С, Н/с |
12 |
14 |
11 |
15 |
13 |
10 |
1,30
|
|||||||
Усилие нажатия на рукоятку У,Н |
300 |
310 |
290 |
300 |
310 |
250 |
1,21 |
|||||||
Физическая нагр узка F, число ударов сердца см3/мин2 |
64 |
67 |
69 |
63 |
68 |
61 |
1,08 |
|||||||
Неудобная рабочая поза П, отн.ед. |
0,59 |
0,62 |
0,60 |
0,64 |
0,63 |
0,53 |
1,16 |
|||||||
Подсистема «машина» |
||||||||||||||
Давление во- здуха в магистрали Р, МПа |
0,50 |
0,48 |
0,52 |
0,54 |
0,48 |
0,50 |
0,95 |
|||||||
Виброскорость инструмента V, дБ |
130 |
127 |
132 |
136 |
127 |
112 |
1,18 |
|||||||
Уровень шума I0, дБ |
83 |
81 |
84 |
82 |
82 |
80 |
1,03 |
|||||||
Время работы с ручной машиной Т, мин |
210 |
230 |
250 |
220 |
240 |
80 |
2,80 |
|||||||
Частота собст-венных колеба-ний f, Гц |
28,5 |
31,7 |
29,0 |
30,8 |
30,2 |
17,9 |
1,70 |
|||||||
Подсистема «окружающая среда» |
||||||||||||||
Освещенность Е, лк |
130 |
128 |
129 |
130 |
127 |
150 |
0,95 |
|||||||
Запыленность N, мг/м3 |
7,1 |
6,8 |
7,6 |
7,2 |
6,9 |
6,0 |
1,09 |
|||||||
Температура окружающей среды t0, С |
12 |
10 |
13 |
11 |
13 |
15 |
0,77 |
|||||||
Барометрическое давление Р0, кПа |
95,3 |
96,7 |
96,0 |
94,7 |
95,6 |
96,0 |
1,08 |
|||||||
Опасные и вредные факторы, которые наибольшим образом способствуют возникновению вибротравматизма, имеют существенные превышения предельно допустимых значений. Основные опасные факторы, формирующие условия труда в подсистеме «машина», приведены в таблице. Используя формулу (1), определяем количественный показатель вредного воздействия каждого из них на условия труда. После соответствующих расчетов получаем значения анализируемых факторов в виде интегрального балла.
Для интегральной оценки условий труда при совокупном воздействии всех факторов используем обобщенный показатель, который определяется из выражения (2): Д=1,70.
Как видно из таблицы, наибольшее неблагоприятное воздействие на вибротравматизм оказывает фактор «время с ручной машиной» (∆yт=2,8).
Фактические значения факторов «освещенность», «температура окружающей среды», «барометрическое давление» меньше, чем нормативные. В данном случае допустим, что неблагоприятное воздействие фактора отсутствует или незначительно влияет на опасность вибротравматизма при ∆уj=1, воздействие фактора будет значимым при 0<∆уj <1.
«Температура окружающей среды» - усугубляющий фактор, влияющий на возникновение вибротравматизма, поскольку нестабильные параметры температуры окружающей среды могут приводить к нарушению механизма терморегуляции, а низкая температура воздуха создает охлаждающий эффект, усиливающий отрицательное воздействие вибрации вследствие сужения кровеносных сосудов.
Согласно формуле (1) определяем интегральный балл влияния каждого фактора подсистемы «окружающая среда» на опасность вибротравмотизма (см. таблицу). Анализ полученных данных показывает, что согласно принятому допущению производственные факторы «освещенность», «запыленность», «барометрическое давление» оказывают незначительное воздействие или неблагоприятное воздействие отсутствует. Основной фактор, который следует считать значимым, - «температура окружающей среды» (∆yt0=0,77)
Совокупное воздействие факторов подсистемы «окружающая среда» определяется в соответствии с формулой (2) и соответствует Д=0,97.
Метод оценки условий труда с использованием интегрального показателя позволяет определить количественную характеристику значимости данного единичного показателя с помощью условий системы численных баллов.
Литература
Злобинского Б.М. Безопасность труда на производстве.-М: Металлургия, 1986.-400с.
Гернет Е.В. Химический анализ воздуха промышленных предприятий.-Л: Химия, 1980.- 440с.
Воронежский государственный технический университет
УДК 541.123.2
В.Н. Чиканов, Ю.Н. Шалимов
ВЗАИМОДЕЙСТВИЕ ГАЛОГЕНИДОВ С ОБЩИМ КАТИОНОМ В РАСПЛАВЕ
На основе анализа двойных диаграмм плавкости рассмотрен ряд факторов, влияющих на взаимодействие галогенидов в системах с общим катионом. Для эвтектических систем проанализированы отклонения температур плавления экспериментальных эвтектик от рассчитанных по уравнению Шредера (идеальныз). В некоторых из них предполагается комплексообразование в расплаве и склонность к образованию твердых растворов
В работах [1-5] приведены критерии, используемые для оценки взаимодействия в солевых системах, но пока нет единого мнения на причины образования того или иного типа диаграмм плавкости. Целью настоящей работы явилось установление факторов, определяющих тип двойных галогенидных диаграмм плавкости с общим катионом.
В таблице 1 приведены типы диаграмм плавкости по данным [6-11]. В большинстве бромидно-хлоридных систем отмечается образование непрерывных твердых растворов без экстремумов или с неглубоким минимумом. Глубина минимума рассчитывалась по соотношению (Тл-пл- Тм) / Тл-пл, где Тл-пл – температура плавления легкоплавкого компонента, Тм - температура плавления, соответствующая минимуму на диаграмме плавкости. Галогениды этих систем имеют сходные кристаллические структуры [12]. Образование эвтектики в системе BiBr3-BiCl3 связано с полиморфизмом у BiBr3, а ограниченных твердых растворов у AlBr3-AlCl3 – с различием кристаллических решеток (AlBr3 – молекулярная, AlCl3 – ионная). В бромидно-иодидных системах наряду с непрерывными твердыми растворами (линии ликвидуса вогнуты к оси абсцисс) образуются твердые растворы с более глубоким минимумом. Система AgBr-AgI содержит ограниченные твердые растворы из-за полиморфизма у AgI. В хлоридно-иодидных системах твердые растворы с очень глубоким минимумом фиксируются в системах, содержащих молекулярные галогениды: TaCl5-TaI5 , NbCl5-NbI5, ZrCl4-ZrI4. В ряде других систем существуют ограниченные твердые растворы. Во фторсодержащих системах твердые растворы не отмечаются.
Максимальная склонность к образованию соединений проявляется в хлоридно-фторидных системах. При взаимодействии галогенидов с кристаллической решеткой типа PbCl2 образуются ионные соединения, исключение составляет SnBr2-SnCl2. Хотя в этой системе и отмечаются ограниченные твердые растворы перетектического типа, но на кривой ликвидуса (75 мол.% SnCl2) существует максимум, который, вероятно, связан с процессами комплексообразования в расплаве (табл.1). Наличию соединений молекулярного типа (SnCl4-SnI4, TiCl4-TiI4) и ниже линии солидуса (GaBr3-GaI3, GaCl3-GaI3) способствуют невысокие температуры плавления компонентов.
Для эвтектических систем авторами проведен анализ отклонений температур плавления эвтектик ( от рассчитанных по уравнению Шредера (таблица 2). Расчет велся по формуле: =(Тл-пл-Тэксп)/(Тл-пл-Трасч), где – относительное отклонение температуры плавления реальной эвтектики от идеальной; Тл-пл – температура плавления легкоплавкого компонента; Тэксп- температура плавления реальной эвтектики; Трасч – температура плавления расчетной эвтектики. Эвтектики фторсодержащих систем с одновалентными катионами близки к идеальным (=1.10.2). В хлоридно-иодидных системах с возможно образование ограниченных твердых растворов. В системах с 1.3 предполагается комплексообразование в расплаве. Величина зависит от интенсивности взаимодействия и температуры плавления галогенидов. Химическое взаимодействие в расплавах систем эвтектического типа отмечается в работах [13-15].
Таблица 1
Типы диаграмм плавкости
Катион |
Анион |
|||||
|
Br-Cl |
Br-I |
Cl-I |
Cl-F |
Br-F |
I-F |
Cs+ |
Тм |
Тм |
То |
Е |
Е |
Е |
Rb+ |
Тм |
Тм |
То |
Е |
Е |
Е |
K+ |
Тм |
Тм |
Ет |
Е |
Е |
Е |
Tl+ |
Тм |
Тм |
Е |
|
|
|
Na+ |
Тм |
Тм |
Ет |
Е |
Е |
Е |
Ag+ |
Тм |
То |
Е |
|
|
|
Li+ |
Тм |
Тм |
е |
Е |
Е |
Е |
Cu+ |
Тм |
Тм |
То |
|
|
|
Ba2+ |
Тм |
|
|
С |
|
|
Sr2+ |
Тм |
Тм |
|
С |
|
|
Pb2+ |
С |
С |
С |
С |
С |
С |
Ca2+ |
|
|
Ес |
С |
|
Ес |
Sn2+ |
То |
С |
|
С |
С |
С |
Mn2+ |
|
|
|
Ес |
|
|
Cd2+ |
Тм |
Тм |
Ет |
Ес |
|
|
Hg2+ |
Тм |
Тм |
То,с |
|
|
|
Mg2+ |
|
|
|
Ес |
|
|
Bi3+ |
Е |
Т |
То,с |
|
|
|
Sb3+ |
|
|
То,с |
|
|
|
Ga3+ |
|
Тм |
Ес |
|
|
|
Al3+ |
То,с |
Тм |
Эс |
|
|
|
U4+ |
|
|
|
С |
|
|
Zr4+ |
Тм |
|
Тм |
|
|
|
Te4+ |
Т |
Тм |
Тм |
|
|
|
Sn4+ |
Тм |
Тм |
С |
|
|
|
Ti4+ |
Тм |
Тм |
С |
|
|
|
Ta5+ |
Т |
Т |
Тм |
С |
|
|
Nb5+ |
Т |
Т |
Тм |
С |
|
|
W5+ |
Т |
|
|
|
|
|
Mo5+ |
|
|
|
С |
|
|
С – соединение; Т – непрерывные твердые растворы; Тм – непрерывные твердые растворы с минимумом; То – ограниченные твердые растворы; То,с - ограниченные твердые растворы, склонные к комплексообразованию в расплаве; Е – системы эвтектического типа; Ет - системы эвтектического типа, склонные к образованию твердых растворов; Ес - системы эвтектического типа, склонные к комплексообразованию в расплаве.
Таким образом, существование твердых растворов в двойных галогенидных диаграммах плавкости с общим катионом обусловлено близостью размеров анионов и сходством кристаллических структур. Наличию соединений способствует более плотная упаковка частиц. При отсутствии этих факторов, в большинстве случаев, образуются системы эвтектического типа.
Таблица 2
Отклонения температуры плавления эвтектики ()
катион |
Br - Cl |
Br - I |
Cl - I |
Cl - F |
Br - F |
I - F |
Ag+ |
|
0.5 |
1.1 |
|
|
|
Cs+ |
|
|
0.9 |
1.2 |
1.3 |
1.3 |
Cu+ |
|
|
0.8 |
|
|
|
K+ |
|
|
0.6 |
1.0 |
1.1 |
1.2 |
Li+ |
|
|
0.9 |
1.1 |
1.3 |
1.1 |
Na+ |
|
|
0.7 |
0.9 |
0.9 |
0.9 |
Rb+ |
|
|
0.6 |
1.1 |
1.2 |
1.2 |
Tl+ |
|
|
0.8 |
|
|
|
|
|
|
|
|
|
|
Ca2+ |
|
|
1.4 |
|
|
2.0 |
Cd2+ |
|
|
0.8 |
1.8 |
|
|
Hg2+ |
|
|
1.7 |
|
|
|
Mg2+ |
|
|
|
8.4 |
|
|
Al3+ |
3.5 |
|
1.8 |
|
|
|
Bi3+ |
0.9 |
|
8.2 |
|
|
|
Ga3+ |
|
|
3.6 |
|
|
|
Sb3+ |
|
|
2.1 |
|
|
|
Литература
1. Федоров П.П., Федоров П.И. // Журн. неорган. химии. 1973. Т.18. С.205.
2. Чиканов Н.Д. // Журн. неорган. химии. 1978. Т.23. С.146.
3. Чиканов Н.Д. // Журн. неорган. химии. 1978. Т.23. С.2596.
4. Чиканов Н.Д. // Журн. неорган. химии. 1981. Т.26. С.752.
5. Витинг Л.М. Высокотемпературные растворы - расплавы. Изд-во Московского университета, 1991. 221 с.
6. Справочник по плавкости систем из безводных неорганических солей. (Составители Воскресенская Н.К. и др.) Том 1. Москва-Ленинград: Изд-во АН СССР, 1961. 845 с.
7. Коршунов Б.Г., Сафонов В.В., Дробот Д.В. Диаграммы плавкости галогенидных систем переходных элементов. М.: Металлургия, 1977. 248 с.
8. Коршунов Б.Г., Сафонов В.В. Галогенидные системы. М.: Металлургия, 1984. 303 с.
9. Коршунов Б.Г., Сафонов В.В. Галогениды. М.: Металлургия, 1991. 288 с.
10. Диаграммы плавкости солевых систем. Справочник (под ред. Посыпайко В.И., Алексеева Е.А.). Часть 3. М.: Металлургия, 1977.
11. Чиканов Н.Д., Моченова И.П. Двойные диаграммы плавкости с участием пентахлоридов, бромидов, иодидов тантала, ниобия и вольфрама // Деп. в ОНИИТЭХИМ г. Черкассы 24.05.85. №627-хп-85.
12. Уэллс А. Структурная неорганическая химия. М.: Мир, 1987. Т.2. 696 с.
13. Воскресенская Н.К. // Известия сектора физико-химического анализа. 1953. Т.23. С.155.
14. Термодинамические свойства расплавов солевых систем. Справочное пособие (отв. ред. Городыский А.В.). Киев: Наук. думка, 1985. 170 с.
15. Волков С.В., Грищенко В.Ф., Делимарский Ю.К. Координационная химия солевых расплавов. Киев: Наукова Думка, 1977. 328 с.
Воронежский государственный технический университет