

Вопрос 95
Инсулин-строение, синтез и секреция. Регуляция синтеза и секреции инсулина. Механизм действия инсулина. Роль инсулина и контринсулярных гормонов (адреналина и глюкагона) в регуляции метаболизма. Изменение гормонального статуса и метаболизма при сахарном диабете. Диабетическая кома.
Инсулин – полипептид, состоящий из двух ППЦ (А и В), соединенных между
собой двумя дисульфидными мостиками.
Может существовать в формах: мономера, димера и гексамера.
Гексамерная структура инсулина стабилизируется ионами Zn2+
Молекула инсулина содержит также внутримолекулярный дисульфидный мостик, соединяющий шестой и одиннадцатый остатки в А-цепи.
Биосинтез инсулина включает обр. 2х неактивных предшественников:
препроинсулина и проинсулина, которые в результате последовательного протеолиза превращаются в активный гормон.
Сигнальный пептид образуется на полирибосомах, проникает в просвет ЭР и
направляет поступление в просвет ЭР растущей ППЦ препроинсулина. После окончания синтеза препроинсулина сигнальный пептид, включающий 24 АК остатка, отщепляется = обр-ся проинсулин
Проинсулин (86 АК остатков) поступает в аппарат Гольджи, где под действием
специфических протеаз расщепляется в нескольких участках = обр. инсулин (51 АК ост)
и С-пептида (31 АК)
Инсулин и С-пептид в эквимолярных количествах включаются в секреторные
гранулы. В гранулах инсулин соединяется с Zn2+, образуя димеры и гексамеры.
Зрелые гранулы сливаются с плазматической мембраной, и инсулин и С-пептид
секретируются во внеклеточную жидкость в результате экзоцитоза. После секреции в кровь олигомеры инсулина распадаются
Распад инсулина происходит под действием инсулиназы (в основном в печени)
РЕГУЛЯЦИЯ СИНТЕЗА И СЕКРЕЦИИ ИНСУЛИНА
Глюкоза – главный регулятор секреции инсулина, а β-клетки – наиболее важные
глюкозо-чувствительные клетки в организме. Глюкоза регулирует экспрессию гена инсулина, а также генов других белков, участвующих в обмене основных энергоносителей.
Действие глюкозы на скорость экспрессии генов инсулина:
Прямое – когда глюкоза непосредственно взаимодействует с
транскрипционными факторами
Вторичное – через влияние на секрецию инсулина и глюкагона.
При стимуляции глюкозой инсулин быстро освобождается из секреторных гранул,
что сопровождается активацией транскрипции мРНК инсулина.
Синтез и секреция инсулина не являются строго сопряжёнными процессами.
Синтез гормона стимулируется глюкозой, а секреция его является Са2+-зависимым процессом и при дефиците Са2+снижается даже в условиях высокой концентрации глюкозы, которая стимулирует синтез инсулина.
На секрецию инсулина влияют другие гормоны:
Адреналин через α2-рецепторы тормозит секрецию инсулина даже на фоне стимуляции глюкозой
β-адренергические агонисты стимулируют синтез (вероятно, в результате повышения концентрации цАМФ)
МЕХАНИЗМ ДЕЙСТВИЯ ИНСУЛИНА:
Он связывается со специфическим гликопротеиновым рецептором на поверхности
клетки-мишени. Рецепторы инсулина обнаружены почти во всех типах клеток, но больше всего их в гепатоцитах и клетках жировой ткани.
Инсулиновый рецептор (IR) постоянно синтезируется и разрушается.
При высокой конц. инсулина в плазме крови (при ожирении) число инсулиновых
рецепторов может уменьшаться, и клетки-мишени становятся менее чувствительными к инсулину, что может быть одной из причин сахарного диабета II типа.
Снижение чувствительности клеток к гормону (десенситизация):
1 механизм включает утрату рецепторов путём их интернализации.
Комплекс инсулин-рецептор захватывается внутрь клетки эндоцитозом. В результате интернализации часть рецепторов подвергается разрушению в лизосомах, а часть возвращается в плазматическую мембрану.
2 механизм – ковалентная модификация рецептора в результате фосфорилир-я
Фосфорилирование IR по остаткам серина и треонина снижает его сродство к инсулину.
Т.к. IR относятся к рецепторам с тирозинкиназной активностью – стимулированное
инсулином аутофосфорилирование β-субъединицы IR по остаткам тирозина приводит к фосфорилированию других внутриклеточных белков – субстратов инсулинового рецептора (IRS) – IRS-1, IRS-2, а также некоторые белки семейства STAT.
IRS-1 - играет главную роль в формировании ответной реакции на инсулиновый
сигнал. Он является фосфопротеином, часть остатков серина, тирозина и треонина фосфорилирована.
При стимуляции инсулином степень фосфорилирования IRS-1 увеличивается и
придаёт ему способность соединяться с другими цитозольными белками, что приводит к активации нескольких сигнальных путей, представляющих каскад реакций активации специфических протеинкиназ.
В результате активации протеинкиназ происходит фосфорилирование ферментов и
факторов транскрипции, что составляет основу многочисленных эффектов инсулина.
Активация инсулином сигнального пути Ras.
Ras-белок – относится к семейству малых ГТФ-связывающих белков.
В неактивном состоянии прикреплён к внутренней поверхности плазматич. мембраны и связан с ГДФ. Стимуляция инсулином приводит к обр. активной ГТФ-связанной формы
Превращение Ras-белка в активную форму происходит при участии семейства
белков, являющихся активаторами протеинкиназ и протеинкиназами
Один из субстратов инсулинового рецептора Shс участвует в образовании
комплекса с небольшим цитозольным белком Grb = образовавшийся комплекс
взаимодействует с Ras-белком. В этот комплекс включаются другие белки: GAP, GEF и SOS. Два последних белка способствуют отделению ГДФ от Ras-белка и присоединению ГТФ. Активированный Ras соединяется с протеинкиназой Raf-1.
ПК Raf-1 в неактивном состоянии находится в цитозоле в соединении с
шаперонами. Активированная Raf-киназа стимулирует каскад реакций фосфорилирования и активации других протеинкиназ – митогенактивируемых протеинкиназ (МАПК).
При участии Raf-1 сначала фосфорилируется и активируется киназа МАПК, которая, в свою очередь, фосфорилирует МАПК.
МАПК фосфорилирует многие цитоплазматические белки: протеинкиназу pp90S6, белки рибосом, фосфолипазу А2, активаторы транскрипции (ПСАТ).

Активация Ras-пути инсулином.
1 – GRB-2/mSOS - цитозольный белок нековалентно присоединяется к
фосфорилированному рецептору инсулина при участии одного из субстратов инсулинововго рецептора - Shс;
2 – образовавшийся комплекс взаимодействует с белком Ras; в этот комплекс
включаются также белки, которые обеспечивают отделение от Ras ГДФ и присоед-е ГТФ;
3 – активированный Ras соединяется с протеинкиназой Raf-1, вследствие чего
происходит активация Raf-1-киназы;
4, 5 – активированная Raf-1-киназа стимулирует каскад реакций
фосфорилирования и активации других протеинкиназ, в частности, МАПКК и МАПК. МАПК фосфорилируют многие цитоплазматические белки и факторы транскрипции. МАПК - митогенактивируемые протеинкиназы.
Активация фосфоинозитол-3-киназы (ФИ-3-киназы): этот фермент катализирует
фосфорилирование ФИ, ФИ-4-фосфата и ФИ-4,5-бисфосфата в положении 3, образуя полифосфоинозитиды: ФИ-3-фосфат, ФИ-3,4-бисфосфат, ФИ-3,4,5-трифосфат, которые в разных клетках стимулируют мобилизацию Са2+и активацию специфических протеинкиназ.
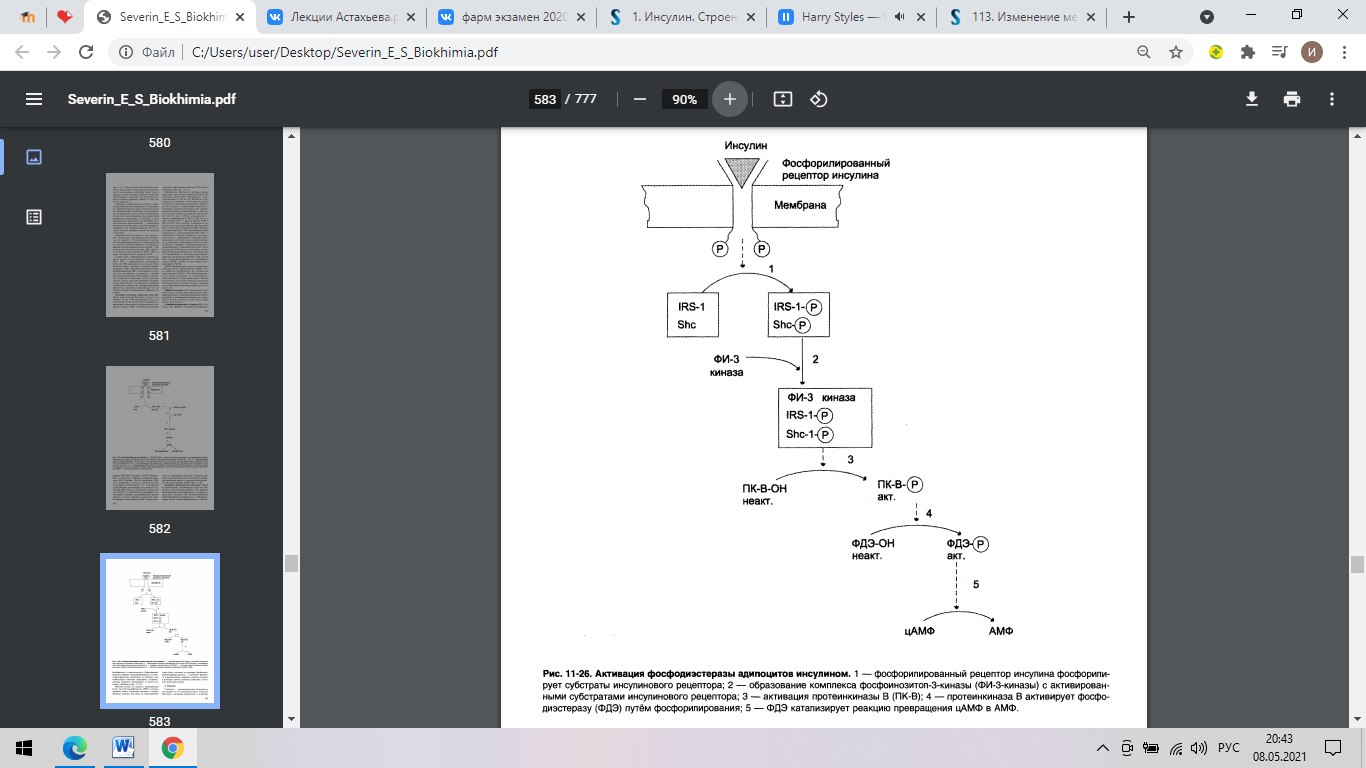
Активация фосфодиэстеразы адипоцитов инсулином.
1 – фосфорилированный рецептор инсулина фосфорилирует субстраты
инсулинового рецептора;
2 – образование комплекса фосфоинозитол-3-киназы (ФИ-3-киназы) с
активированными субстратами инсулинового рецептора;
3 – активация протеинкиназы В (ПК-В);
4 – протеинкиназа В активирует фосфодиэстеразу (ФДЭ) путём фосфорилирования
5 – ФДЭ катализирует реакцию превращения цАМФ в АМФ.
Активация гликогенсинтазы инсулином. Одной из протеинкиназ, активируемых
через путь Ras. является протеинкиназа pp90S6. Этот фермент фосфорилирует протеинфосфатазу, связанную с гранулами гликогена.
При фосфорилировании протеинфосфатаза активируется и дефосфорилирует
киназу гликогенфосфорилазы, гликогенфосфорилазу и гликогенсинтазу.
Дефосфорилированные формы киназыфосфорилазы и гликогенфосфорилазы неактивны, вследствие чего мобилизация гликогена замедляется.
Гликогенсинтаза, напротив, активируется, и синтез гликогена ускоряется.
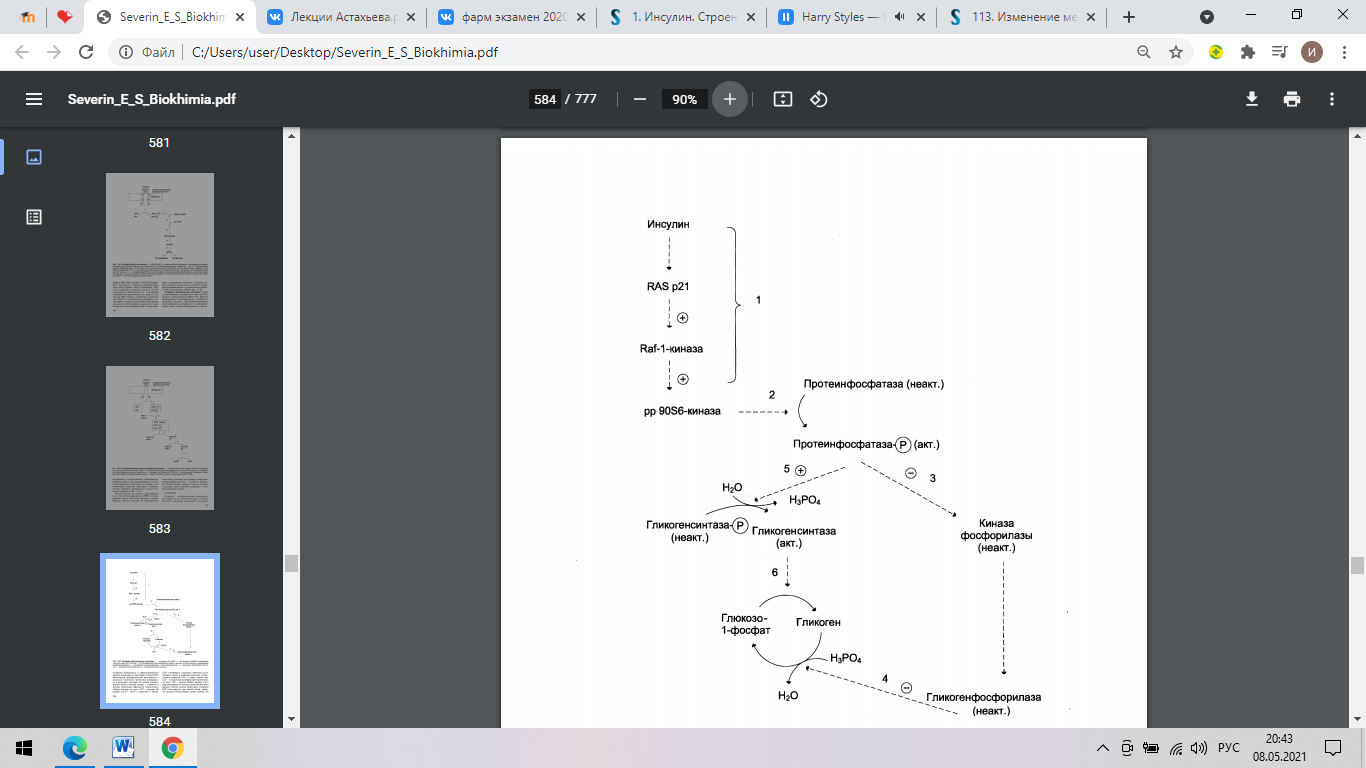
Активация гликогенсинтазы инсулином.
1 – активация пути RAS;
2 – протеинкиназа рр90S6, активируемая инсулином через путь RAS,
фосфорилирует протеинфосфатазу гранул гликогена, которая включает каскад реакций дефосфорилирования;
3 – инактивация киназыфосфорилазы и гликогенфосфорилазы;
4 – торможение мобилизации гликогена;
5 – активация гликогенсинтазы;
6 – стимуляция синтеза гликогена.
РОЛЬ ИНСУЛИНА И КОНТРИНСУЛЯРНЫХ ГОРМОНОВ (АДРЕНАЛИНА И ГЛЮКАГОНА) В РЕГУЛЯЦИИ МЕТАБОЛИЗМА.
Основную роль в поддержании энергетического гомеостаза играют
гормоны инсулин и глюкагон, а также другие контринсулярные гормоны – адреналин, кортизол, йодтиронины и соматотропин.
Инсулин и глюкагон играют главную роль в регуляции метаболизма при смене
абсорбтивного и постабсорбтивного периодов и при голодании.
Абсорбтивный период характеризуется временным повышением конц. глюкозы,
аминокислот и жиров в плазме крови. Клетки поджелудочной железы отвечают на это повышение усилением секреции инсулина и снижением секреции глюкагона.
Увеличение отношения инсулин/глюкагон вызывает ускорение использования
метаболитов для запасания энергоносителей: происходит синтез гликогена, жиров и белков. Режим запасания включается после приёма пищи и сменяется режимом мобилизации запасов после завершения пищеварения. Тип метаболитов, которые потребляются, депонируются и экспортируются, зависит от типа ткани.
Главные органы, связанные с изменениями потока метаболитов при смене режимов
мобилизации и запасания энергоносителей, - печень, жировая ткань и мышцы.
ИЗМЕНЕНИЕ ГОРМОНАЛЬНОГО СТАТУСА И МЕТАБОЛИЗМА ПРИ САХАРНОМ ДИАБЕТЕ
. Диабетическая кома.
Сахарный диабет – заболевание, возникающее вследствие абсолютного или
относительного дефицита инсулина.
Диабет I типа – инсулинзависимый – заболевание, вызываемое разрушением
р-клеток островков Лангерханса поджелудочной железы.
Диабет II типа – инсулиннезависимый – общее название нескольких
заболеваний, развивающихся в результате относительного дефицита инсулина, возникающего вследствие нарушения секреции инсулина, нарушения превращения проинсулина в инсулин, повышения скорости катаболизма инсулина, а также повреждения механизмов передачи инсулинового сигнала в клетки-мишени (например, дефекта рецептора инсулина, повреждения внутриклеточных посредников
инсулинового сигнала и др.)
При сахарном диабете, как правило, соотношение инсулин/глюкагон снижено.
При этом ослабевает стимуляция процессов депонирования гликогена и жиров, и усиливается мобилизация запасов энергоносителей.
Печень, мышцы и жировая ткань даже после приёма пищи функционируют в
режиме постабсорбтивного состояния. Для всех форм диабета характерна гипергликемия
После приёма пищи концентрация глюкозы может достигать 300-500 мг/дл и
сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе.
При снижении инсулинглюкагонового индекса активируется глюконеогенез из
аминокислот, глицерола и лактата.
Повышение концентрации глюкозы в крови при сахарном диабете превышает
концентрационный почечный порог, что становится причиной выделения глюкозы
с мочой (глюкозурия).
К характерным признакам сахарного диабета относят также повышение
концентрации в крови кетоновых тел – кетонемия.
При низком соотношении инсулин/глюкагон жиры не депонируются, а ускоряется
их катаболизм. Печень захватывает жирные кислоты, окисляет их до ацетил-КоА, который, в свою очередь, превращается в β-гидроксимасляную и ацетоуксусную кислоты. В тканях ацетоацетат частично декарбоксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии. Увеличение концентрации кетоновых тел в крови приводит к кетонурии.
Накопление кетоновых тел снижает буферную ёмкость крови вызывает ацидоз.
При сахарном диабете дефицит инсулина приводит к снижению скорости синтеза
белков в организме и усилению распада белков.Образующийся при этом аммиак вступает в орнитиновый цикл, что приводит к увеличению концентрации мочевины в крови и моче – азотемия и азотурия.
Выделение мочи у больных возрастает в несколько раз и в некоторых случаях
достигает 8-9 л в сутки, но чаще не превышает 3-4 л - полиурия. Потеря воды вызывает постоянную жажду – полидипсия.
Диабетическая кома – резкое нарушение всех функций организма с
нарушением сознания. Она развивается медленно, в течение нескольких дней, но иногда может возникнуть возникнуть и в течение нескольких нескольких часов.
Основные предшественники диабетической комы – ацидоз и дегидратация тканей.
Параллельно кетоацидозу при декомпенсации диабета развивается нарушение
водно-электролитного обмена. В его основе лежит гипергликемия, сопровождающаяся повышением осмотического давления в сосудистом русле.
Для сохранения осмолярности начинается компенсаторное перемещение жидкости
из клеток и внеклеточного пространства в сосудистое русло, что ведёт к потере тканями воды и электролитов – развиваются тяжёлая клеточная дегидратация и дефицит внутриклеточных ионов (прежде всего К+), затем возникает общая дегидратация.
Это приводит к снижению периферического кровообращения, уменьшению
мозгового и почечного кровотока и гипоксии.
Первыми признаками могут быть тошнота, рвота, заторможенность, АД снижено.
Коматозные состояния при сахарном диабете проявляются в 3х формах:
Для кетоацидотической комы характерны выраженный дефицит инсулина,
кетоацидоз, полиурия, полидипсия. Гипергликемия (20 — 30 ммоль/л), обусловленная инсулиновой недостаточностью, сопровождается большими потерями жидкости и электролитов, дегидратацией и гиперосмоляльностью плазмы. Общая концентрация кетоновых тел достигает 100 мг/дл и выше.
При гиперосмолярной коме наблюдают чрезвычайно высокие уровни глюкозы в
плазме крови, полиурию, полидипсию, всегда проявляется тяжёлая дегидратация. Предполагают, что у большинства больных гипергликемия обусловлена сопутствующим нарушением функции почек. Кетоновые тела в сыворотке крови обычно не определяются.
При лактоацидотической коме преобладают гипотония, снижение
периферического кровообращения, гипоксия тканей, приводящая к смещению метаболизма в сторону анаэробного гликолиза, что обусловливает повышение концентрации молочной кислоты в крови (лактоацидоз)
Вопрос 96
Гормоны щитовидной железы. Регуляция синтеза и секреции йодтиронинов и их влияние на метаболизм и функции организма. Изменение метаболизма при гипо- и гипертиреозе. Причины и проявления эндемического зоба.
В щитовидной железе синтезируются гормоны – йодированные производные
тирозина (объеденены общим названием йодтиронины)
К ним относят: трийодтиронин (Т3), тетрайодтиронин (Т4) или тироксин.
1. Биосинтез йодтиронинов: они синтезируются в составе белка тиреоглобулина
(Тг) в фолликулах щитовидной железы.
Тиреоглобулин – гликопротеин, содержащий 115 остатков тирозина.
Он синтезируется на рибосомах шероховатого ЭР в виде претиреоглобулина,
затем переносится в цистерны ЭР, где происходит формирование вторичной и третичной структуры, включая процессы гликозилирования. Из цистерн ЭР поступает в аппарат Гольджи, включается в состав секреторных гранул и секретируется во внеклеточный коллоид, где происходит йодирование остатков тирозина и образование йодтиронинов.
2. Транспорт йода в клетки щитовид. железы: йод в виде органических и
неорганических соединений поступает в ЖКТ с пищей и питьевой водой. Транспорт йодида в клетки щитовидной железы – энергозависимый процесс и происходит при участии специального транспортного йодид-переносящего белка, работа которого сопряжена с Na+, K+-АТФ-азой
3. Окисление йода: окисление I- в I+ происходит при участии гемсодержащей
тиреопероксидазы и H2O2 в качестве окислителя.
4. Йодирование тирозина: окисленный йод взаимодействует с остатками тирозина
в молекуле тиреоглобулина. Реакция катализируется тиреопероксидазой.
5. Образование йодтиронинов: под действием тиреопероксидазы оксиленный йод
реагирует с остатками тирозина с образованием монойодтирозинов (МИТ) и дийодтирозинов (ДИТ). Две молекулы ДИТ конденсируются с образованием йодтиронина Т4, а МИТ и ДИТ – с образованием йодтиронина Т3. Йодтиреоглобулин транспортируется из коллоида в фолликулярную клетку путем эндоцитоза и гидролизуется ферментами лизосом с высвобождением Т3 и Т4.
Транспорт и метаболизм йодтиронинов: большая часть Т3 и Т4 находятся в
организме вне щитовидной железы – они циркулируют в крови в связанной форме в комплексе с белками: тироксинсвязывающим глобулином (ТСГ) и тироксинсвязывающим преальбумином (ТСПА). И лишь малая частьТ4 и Т3 находятся в крови в свободной форме.
В периферических тканях в результате дейодирования части Т4 по пятому
углеродному атому образуется «реверсивная» форма Т3, которая почти лишена биологической активности. Другие пути метаболизма йодтиронинов включают полное дейодирование, дезаминирование или декарбоксилирование.

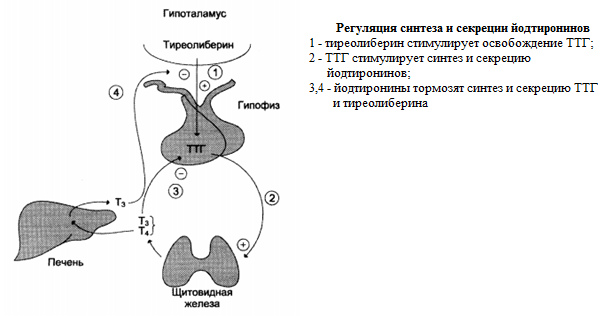
Регуляция синтеза и секреции йодтиронинов: скорость синтеза и секреции
йодтиронинов регулируются гипоталамо-гипофизарной системой по механизму обратной отрицательной связи.
Стимулом для повышения секреции тиреолиберина и как следствие синтеза тиреотропного гормона служит снижение концентрации йодтиронинов в крови.
Йодтиронины тормозят синтез и секрецию ТТГ и тиреолиберина.
Биологические функции: в печени йодтиронины ускоряют гликолиз, синтез
холестерола и синтез желчных кислот.
В печени и жировой ткани повышает чувствительность клеток к действию адреналина и косвенно стимулирует липолиз мобилизацию гликогена
Т3 увеличивает в мышцах потребление глюкозы, стимулирует синтез белков и увеличение мышечной массы,
Йодтиронины также участвуют в формировании ответной реакции на охлаждение
увеличением теплопродукции, повышая чувствительность симпатической нервной системы к норадреналину и стимулируя секрецию норадреналина.
ЗАБОЛЕВАНИЯ ЩИТОВИДНОЙ ЖЕЛЕЗЫ
Гипотиреоз у новорожденных приводит к развитию кретинизма, который
проявляется множественными врожденными нарушениями и тяжелой необратимой задержкой умственного развития.
Гипотиреоз развивается вследствие недостаточности йодтиронинов. Обычно
связан с недостаточностью функции щитовидной железы, но может возникать и при заболеваниях гипофиза и гипоталамуса.
Микседема – наиболее тяжелая форма гипотиреоза, сопровождается слизистым
отеком кожи и подкожной клетчатки. Отечность обусловлена избыточным накоплением гликозаминогликанов и воды. Характерные проявления: снижение частоты сердечных сокращений, вялость, сонливость, непереносимость холода, сухость кожи.
Эндемический зоб – встречается у людей, живущих в районах, где содержание
йода в воде и почве недостаточно. Если поступление йода в организм снижается, то уменьшается продукция йодтиронинов, что приводит к усилению секреции ТТГ, под влиянием которого происходит компенсаторное увеличение размеров щитовидной железы, но продукция йодтиронинов при этом не увеличивается.
Гипертиреоз возникает вследствие повышенной продукции йодтиронинов.
Диффузный токсический зоб (базедова болезнь) – увеличение размеров
щитовидной железы, повышение концентрации йодтиронинов в 2-5 раз и развитие тиреотоксикоза. Признаки тиреотоксикоза: увеличение основного обмена, учащение сердцебиений, мышечная слабость, снижение массы тела (несмотря на повышенный аппетит), потливость, тремор, экзофтальм (пучеглазие).
Причины гипертиреоза: развитие опухоли, тиреоидит, избыточное поступление йода и йодсодержащих препаратов, аутоиммунные реакции.