

921
.pdfВид анализа |
Тип реакции |
Кислотно-основное титрование |
Реакция нейтрализации |
Комплексонометрическое |
Образование комплексного соединения |
титрование |
|
Окислительно-восстановитель- |
Реакция окисления-восстановления |
ное титрование |
|
Осадительное титрование |
Образование нерастворимого соединения |
7.3.Инструментальные методы анализа
Кинструментальным методам анализа относят: 1) физические – методы, в которых с помощью приборов измеряют какое-либо свойство вещества без проведения химической реакции и 2) физикохимические – методы, в которых за ходом химической реакции следят
спомощью приборов, определяя, как в ходе реакции изменяется то или иное свойство раствора. В табл.32 приведены наиболее распространенные инструментальные методы анализа.
Основные инструментальные методы анализа |
|
Таблица 32 |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||
Метод |
Измеряемая величина |
Закон, который лежит |
|||||||||||||
|
|
в основе метода |
|||||||||||||
1 |
2 |
|
|
|
3 |
|
|
|
|
|
|
|
|||
Электрохимические |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Потенциометрический |
Электродный потенциал, |
Уравнение Нернста |
|||||||||||||
|
ЭДС |
E E |
0 |
|
|
RT |
ln |
aox |
|
||||||
|
|
|
|
nF |
ared |
||||||||||
|
|
|
|
|
|
|
|
||||||||
Вольтамперометрический |
Зависимость силы тока от |
Закон Ома I |
U |
|
|||||||||||
(полярографический) |
величины напряжения |
R |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||
Кулонометрический |
Количество электричества |
Закон Фарадея |
|||||||||||||
|
|
|
m |
ЭIt |
|
||||||||||
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
F |
|
|
|
|
|
|
|
Кондуктометрический |
Электрическая |
10 3 |
|
A |
|
iCi |
|||||||||
|
проводимость |
|
|
|
|
d |
|
|
|
|
|
|
|||
|
Масса вещества, |
Закон Фарадея |
|||||||||||||
Электрогравиметрия |
образовавшаяся в |
|
m |
ЭIt |
|
||||||||||
|
процессе |
|
|
|
|
|
|
F |
|
|
|
|
|
|
|
|
электролиза |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Окончание табл. 32 |
|||||||||||||
1 |
2 |
|
|
|
3 |
|
|
|
|
|
|
|
|||
Оптические |
|
|
|
|
|
|
|
|
|
|
|
Эмиссионная спектроскопия |
Спектр излучения |
Эмпирическое |
|||||||||
|
вещества при сжигании |
уравнение Ломакина |
|||||||||
|
пробы |
I a cb |
|||||||||
Люминесцентный |
Спектр люминесценции |
I = k∙c |
|||||||||
(флуоресцентный) |
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
Абсорбционная спектро- |
Спектр поглощения |
Закон Бугера - |
|||||||||
скопия (визуальная |
электромагнитного |
Ламберта Бера |
|||||||||
колориметрия, |
излучения веществом |
D lg |
I0 |
|
c l |
||||||
фотоколоримет-рия и |
|
|
|
|
I |
|
|
|
|
||
спектрофотометрия) |
|
|
|
|
|
|
|
|
|
|
|
Турбидиметрия |
Снижение интенсивности |
S lg |
I0 |
|
k b N |
||||||
|
света из-за его поглощения |
|
|||||||||
|
|
|
|
||||||||
|
взвешенными частицами |
|
I |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Нефелометрия |
Интенсивностьрассеянного |
Закон Релея |
|||||||||
|
света |
I Ka c I0 |
|||||||||
Рефрактометрия |
Показатель преломления |
c |
|
n n0 |
|
||||||
|
|
|
|
|
|
F |
|||||
|
|
|
|
|
|
|
|
|
|||
Поляриметрия |
Изменение угла вращения |
c |
100 |
|
|||||||
|
плоскости поляризации |
|
|
|
|
D20 l |
|||||
|
света оптически |
|
|
|
|
|
|
|
|
|
|
|
активными веществами |
|
|
|
|
|
|
|
|
|
|
Электрохимические методы анализа основаны на изучении и использовании процессов, проходящих на поверхности электрода или в приэлектродном пространстве.
Оптические методы основаны на взаимодействии электромагнитного излучения с веществом: его поглощении, отражении, рассеивании, преломлении, испускании.
Отдельнуюгруппусоставляютхроматографическиеметоды(табл.33). Это не только методы анализа, но и методы разделения веществ, основанные на распределении компонентов между двумя фазами: подвижной и неподвижной. Неподвижной фазой служит твердое вещество или пленка вязкой нелетучей жидкости, нанесенная на твердую инертную поверхность. Подвижная фаза – легколетучая жидкость или газ, проходящий через неподвижную фазу. Компоненты, слабо взаимодействующие с неподвижной фазой, быстро продвигаются вместе с подвижной фазой, а компоненты, хорошо взаимодействующие с неподвижной фазой, продвигаются
медленно. Компоненты сначала разделяют, а потом определяют.
Таблица 33
Классификация хроматографических методов
|
Вид хроматографии |
Неподвижная |
Подвижная |
На чем основан метод |
|
|
фаза |
фаза |
|
|
Газовая |
|
|
|
|
Адсорбционная |
Тв. |
Газ |
Различная способность |
|
|
|
|
компонентов к адсорб- |
|
|
|
|
ции на поверхности |
|
|
|
|
неподвижной фазы |
|
Газожидкостная |
Ж. |
Газ |
Различная раствори- |
Колоночная |
(распределительная) |
|
|
мость компонентов в |
|
|
|
неподвижной фазе |
|
Жидкостная |
|
|
|
|
Распределительная |
Ж. |
Ж. |
Разные коэффициенты |
|
|
|
|
распределения компо- |
|
|
|
|
|
|
|
|
|
|
нентов между двумя |
|
|
|
|
несмешивающимися |
|
|
|
|
жидкостями |
|
Ионообменная |
Тв. ионо- |
Ж. |
Способность к |
|
|
обменная |
|
ионному обмену |
|
|
смола |
|
|
Плоскостная |
Бумажная |
Хроматогра- |
Ж. |
Различная способность |
|
фическая |
|
компонентов к |
|
|
бумага |
|
адсорбции |
|
Тонкослойная |
Сорбент, |
Ж. |
|
|
|
нанесенный |
|
|
|
|
тонким слоем |
|
|
|
|
|
на пластину |
|
|
8.ОСНОВЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
8.1.Определение теплового эффекта химических реакций
Расчеты тепловых эффектов химических реакций базируются на законе Гесса и следствиях из него.
Закон Гесса: тепловой эффект химической реакции (т.е. изменение энтальпии или внутренней энергии системы в результате реакции) зависит только от начального и конечного состояния участвующих в реакции веществ и не зависит от промежуточных стадий процесса.
H х.р = H прод – H исх. в-в ,
где H х.р – тепловой эффект химической реакции;
H прод и H исх. в-в – сумма энтальпий продуктов реакции и исходных веществ, соответственно.
Следствие 1: стандартное изменение энтальпии химической реакции равно разности между суммой стандартных энтальпий
образования продуктов реакции и |
суммой стандартных энтальпий |
|
образования исходных веществ. |
|
|
H х.р |
= Hо (обр) прод |
– Hо(обр) исх. в-в , |
где Hо(обр) прод и |
Hо(обр) исх. в-в |
– сумма стандартных энтальпий |
образования продуктов реакции и исходных веществ соответственно, кДж/моль.
Следствие 2: тепловой эффект (изменение энтальпии) химической реакции равен разности энтальпий сгорания продуктов реакции и исходных веществ, взятой с противоположным знаком.
H х.р |
= |
Hо (сгор) |
исх. в-в |
– Hо(сгор) прод , |
где Hо(сгор) прод |
и |
Hо(сгор) |
исх. в-в |
– сумма теплот (энтальпий) |
сгорания продуктов реакции и исходных веществ соответственно, кДж/моль.
Для уравнения химической реакции, записанного в общем виде: aA + bB = cC +dD
эти следствия запишутся так:
Следствие (1):
H х.р = c Hо(обр.)C + d Hо(обр.)D – a Hо (обр.).A – b Hо (обр.). B,
Следствие(2):
H х.р = a Hо (сгор.).A +b Hо (сгор.). B – c Hо(сгор.)C – d Hо(сгор.)D.
8.2. Определение направления протекания химической реакции
Для определения направления протекания химических процессов используется характер изменения изобарно-изотермического потенциала (свободной энергии, или энергии Гиббса) системы G, рассчитываемого по уравнению
G х.р = Gо прод – Gо исх. в-в ,
где Gопрод и Gоисх. в-в – суммы стандартных значений энергий Гиббса продуктов реакции и исходных веществ соответственно, кДж/моль.
Если G х.р 0, то процесс возможен и протекает самопроизвольно. Если G х.р 0, то процесс термодинамически невозможен при
данных условиях (но возможен в направлении справа налево).
Если G х.р = 0, то система находится в состоянии химического равновесия.
Вычислить G можно по уравнению
G = H – Т S,
где H – изменение энтальпии при химической реакции; Т – температура, К;
S – изменение энтропии при химической реакции.
Hх.р = Hо(обр.) прод – Hо(обр.) исх. в-в ,
Sх.р= Sопрод – Sоисх. в-в ,
где Hо(обр. )прод и Hо(обр.) исх. в-в – значения стандартных энтальпий образования продуктов реакции и исходных веществ соответственно, кДж/моль,
Sопрод и Sоисх. в-в – стандартные значения энтропий продуктов реакции и исходных веществ соответственно, Дж/моль.
Для уравнения химической реакции, записанного в общем виде: aA + bB = cC +dD
H х.р, S х.р и G х.р рассчитываются так:
Hх.р = c Hо(обр.)C + d Hо(обр.)D – a Hо(обр.)A – b Hо(обр.)B,
Sх.р = cSоC + dSоD –aSоA – bSоB,
Gх.р = c GоC + d GоD – a GоA – b GоB.
9.ХИМИЧЕСКАЯ КИНЕТИКА И КАТАЛИЗ
9.1.Зависимость скорости химической реакции от концентрации
Зависимость скорости химической реакции от концентрации реагирующих веществ выражает закон действующих масс, сформулированный в 1867 г. Гульбергом и Вааге:
Скорость химической реакции при постоянной температуре пропорциональна произведению концентраций реагирующих веществ, возведенных в степень их стехиометрических коэффициентов (т.е.
коэффициентов химического уравнения).
Для уравнения в общем виде: aA + bB = cC +dD скорости прямой и обратной реакций записываются так:
υпр = k1 СА а СBb ,
υобр = k2 СC c СDd,
где k1 и k2 – константы скорости прямой и обратной реакций соответственно, не зависящие от концентрации реагирующих веществ;
СА, СB – концентрации исходных веществ;
СC, СD – концентрации продуктов реакции;
а, b, с, d – стехиометрические коэффициенты в уравнении.
9.2. Зависимость скорости химической реакции от температуры
Зависимость скорости химической реакции от температуры можно определить с помощью эмпирического правила Вант-Гоффа:
При повышении температуры на каждые десять градусов скорость большинства химических реакций возрастает в 2 4 раза:
|
|
|
T2 T1 |
|
T |
T |
|
10 |
, |
2 |
1 |
|
|
|
где υ(Т2) и υ(Т1) – скорости химической реакции при температурах Т2 и Т1, соответственно;- температурный коэффициент скорости реакции (от 2 до 4).
Более точно зависимость скорости (точнее константы скорости k) реакции от температуры может быть рассчитана с помощью
уравнения Аррениуса:
k = A e –Ea/RT,
где A – величина, постоянная для данной реакции, приблизительно равна числу соударений реагирующих частиц в единице объема в единицу времени;
e – основание натурального логарифма, приблизительно равное 2,718; R – универсальная газовая константа, равная 8,314 Дж/моль К;
Т – температура, К;
Ea – энергия активации, кДж/моль.
9.3. Каталитические реакции
Катализаторами называются вещества, которые влияют на скорость реакции, но сами в ходе реакции не расходуются.
Катализаторы не влияют на термодинамику реакции (не изменяют G, H), т.е. если G > 0, то в присутствии катализатора она не станет самопроизвольной. Если реакция эндотермическая ( Н > 0), то в присутствии катализатора она не станет экзотермической.
Катализаторы не влияют на смещение химического равновесия, они могут только ускорять наступление химического равновесия. Катализатор увеличивает константу скорости химической реакции, следовательно, в одинаковой степени влияет на константу скорости прямой и обратной реакций. Катализаторы уменьшают полную энергию активации процесса. Активированный комплекс с
катализатором имеет меньшую энергию, чем |
комплекс без |
катализатора, следовательно, Еа реакции > Еа |
каталитической |
реакции. |
|
Катализаторы, которые находятся в одной фазе с реагирующими веществами, называются гомогенными. Катализатор, представляющий отдельную фазу, на поверхности которой идет каталитическая реакция, называется гетерогенным. Механизм гетерогенного катализа всегда сложнее гомогенного, так как в нем обязательно есть стадия адсорбции, то есть взаимодействие реагента с поверхностью катализатора.
10. ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Химическая система находится в состоянии химического равновесия, если скорость прямой реакции равна скорости обратной реакции, концентрации всех веществ, участвующие в ходе этой реакции, постоянны и не изменяются во времени; энергия системы минимальна и не изменяется (ΔG = 0).
10.1. Константа химического равновесия
Для уравнения химической реакции, записанном в общем виде: aA + bB = cC +dD,
константа химического равновесия K определяется по уравнению
|
c |
d |
|
K |
[C] |
[D] |
, |
a |
b |
||
|
[A] |
[B] |
|
где А , В , С , D - равновесные концентрации реагирующих веществ, моль/л.
а, b, с, d – стехиометрические коэффициенты в уравнении.
0.2. Смещение химического равновесия
Направление смещения химического равновесия при изменении внешних условий (температуры, давления, концентраций)
определяется принципом Ле-Шателье: если на систему, находящуюся в состоянии равновесия, производится какое-либо внешнее воздействие, то равновесие сместится в сторону ослабления данного воздействия.
1) Повышение температуры приводит к смещению равновесия в сторону эндотермической реакции (протекающей с поглощением тепла; H х.р 0) и наоборот, понижение температуры приводит к смещению равновесия в сторону экзотермической реакции (протекающей с выделением тепла; H х.р 0).
2)Повышение давления в газовой смеси смещает равновесие в сторону уменьшения числа молей газообразных веществ.
3)Удаление из системы хотя бы одного из продуктов реакции смещает равновесие в сторону прямой реакции, а уменьшение концентрации исходных веществ – в сторону обратной реакции. (При повышении концентрации реагирующих веществ взаимосвязь обратная.)
11.ОБЩИЕ СВОЙСТВА РАСТВОРОВ
11.1. Законы идеальных растворов
Идеальным называется раствор, между компонентами которого нет химического взаимодействия, а силы межмолекулярного взаимодействия примерно одинаковы. Для идеальных растворов отсутствуют тепловые и объемные эффекты растворения. Ближе всего по свойствам к идеальным растворам разбавленные растворы неэлектролитов.
11.1.1. Изменение давления пара растворителя в зависимости от концентрации растворенного вещества (I закон Рауля)
Уменьшение давления насыщенного пара растворителя над раствором прямо пропорционально мольной доле нелетучего растворенного компонента P P0 P P0 xв ва ,
где P – давление насыщенного пара растворителя над раствором; Р0 – давление насыщенного пара над чистым растворителем; хв-ва – мольная доля вещества.
11.1.2.Понижение температуры замерзания раствора
иповышение температуры кипения раствора (II закон Рауля)
Понижение температуры замерзания раствора по сравнению с температурой замерзания чистого растворителя прямо пропорционально моляльной концентрации растворенного вещества.
Tзам K Cm.
Повышение температуры кипения раствора по сравнению с температурой кипения чистого растворителя прямо пропорционально моляльной концентрации растворенного вещества.
Tкип E Cm,
где Сm – моляльная концентрация вещества, моль/кг растворителя; К – криоскопическая константа растворителя; Е – эбуллиоскопическая константа растворителя.
Криоскопическая и эбуллиоскопическая константы характеризуют только растворитель и не зависят от химической природы растворенного вещества. Значения криоскопических и эбуллиоскопических констант приведены в табл. 34.
Таблица 34
Криоскопические и эбуллиоскопические константы некоторых растворителей
Константа |
Н2О |
С2Н5ОН |
С6Н6 |
ССl4 |
К град∙моль/кг |
1,85 |
1,99 |
5,12 |
29,8 |
Е град∙моль/кг |
0,52 |
1,22 |
2,53 |
5,02 |
11.1.3. Осмотическое давление (закон Вант-Гоффа)
Осмотическое давление раствора прямо пропорционально молярной концентрации раствора и температуре этого раствора.
Pосм См R T ,
где Росм – осмотическое давление, кПа; См – молярная концентрация, моль/дм3 , или моль/л,
R 8,314Дж/моль∙К – универсальная газовая постоянная;
Т– абсолютная температура, К.
11.2.Отклонение от законов идеальных растворов

Для растворов электролитов наблюдаются положительные отклонения от законов идеальных растворов, так как в растворе молекулы электролита распадаются на ионы. Количество частиц в растворе увеличивается, меняется давление насыщенных паров растворителя, осмотическое давление и т.п. Для того чтобы учесть влияние электролитической диссоциации на свойства раствора, вводят изотонический коэффициент. Он показывает, во сколько раз отличаются свойства реального раствора от идеального.
i Pреал Tзам(реал) Росм(реал) .
Ридеал Tзам(идеал) Росм(идеал)
Для концентрированных растворов могут наблюдаться как положительные, так и отрицательные отклонения от законов идеальных растворов. Это вызвано различием в силах межмолекулярного взаимодействия между однородными и разнородными частицами. Для учета взаимного влияния ионов друг на друга ввели понятие активности (кажущейся концентрации) ионов и молекул в реальных растворах. Активность связана с молярной концентрацией формулой
a c ,
где а – активность ионов или молекул, моль/дм3; γ – коэффициент активности (безразмерная величина); с – концентрация ионов или молекул, моль/дм3.
Коэффициент активности зависит от количества присутствующих в растворе ионов и вычисляется по уравнению Дебая – Хюккеля:
lg AzAzB 
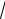 I ,
I ,
где А – постоянная, зависящая от природы растворителя; zA и zB – заряды катиона и аниона;
I – ионная сила раствора.
Ионная сила раствора зависит от природы и концентрации всех ионов, присутствующих в растворе и вычисляется по формуле
I 12 cizi2 ,
где сi – концентрация каждого иона; zi – заряд иона.