

Strukov_A_I_Serov_V_V_Patanatomia_Uchebnik_20
.pdfтоцитах, мышечных и нервных клетках, а также в клетках коры надпочечников.
Микроскопическая картина: паренхиматозные клетки увеличены в объеме, цитоплазма их заполнена вакуолями, содержащими прозрачную жидкость. Ядро смещается на периферию, иногда вакуолизируется или сморщивается. Прогрессирование этих изменений приводит к распаду ультраструктур клетки и переполнению клетки водой. Клетка превращается в заполненные жидкостью баллоны или в огромную вакуоль, в которой плавает пузырьковидное ядро. Такие изменения клетки, которые по существу являются выражениемфокального колликвационного некроза называют баллонной дистрофией.
Внешний вид органов и тканей мало изменяется при гидропическои дистрофии, она обнаруживается обычно под микроскопом.
Механизм развития гидропическои дистрофии сложен и отражает нарушения водноэлектролитного и белкового обмена, ведущие к изменению коллоидно-осмотического давления в клетке. Большую роль играет нарушение проницаемости мембран клетки, сопровождающееся их распадом. Это ведет к закислению цитоплазмы, активации гидролитических ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды.
Причины развития гидропической дистрофии в разных органах неоднозначны. В почках - это повреждение гломерулярного фильтра (гломерулонефрит, амилоидоз, сахарный диабет), что ведет к гиперфильтрации и недостаточности ферментной системы базального лабиринта нефроцитов, в норме обеспечивающей реабсорбцию воды; поэтому гидропическая дистрофия нефроцитов так характерна для нефротического синдрома.
В печени гидропическая дистрофия возникает при вирусном и токсическом гепатитах (рис. 28) и нередко является причиной печеночной недостаточности. Причиной гидропическои дистрофии эпидермисаможет быть инфекция (оспа), отек кожи различного механизма. Вакуолизация цитоплазмы может быть проявлением физиологической деятельности клетки, что отмечается, например, в ганглиозных клетках центральной и периферической нервной системы.
Исход гидропической дистрофии, как правило, неблагоприятный; она завершается фокальным или тотальным некрозом клетки. Поэтому функция органов и тканей при гидропической дистрофии резко страдает.
Роговая дистрофия
Роговая дистрофия, или патологическое ороговение, характеризуется избыточным образованием рогового вещества в ороговевающем эпителии (гиперкератоз, ихтиоз) или образованием рогового вещества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках, или лейкоплакия; образование «раковых жемчужин» в плоскоклеточном раке). Процесс может быть местным или распространенным.
61

Рис.
а - микроскопическая картина; вакуолизация гепатоцитов; б - электронограмма: расширение канальцев эндоплазматической сети и образование вакуолей (В), заполненных хлопьевидным содержимым. Мембраны, ограничивающие вакуоли, почти полностью лишены рибосом. Вакуоли сдавливают расположенные между ними митохондрии (М), часть которых подвергается деструкции; Я - ядро гепатоцита. х18 000
Причины роговой дистрофии разнообразны: нарушение развития кожи, хроническое воспаление, вирусные инфекции, авитаминозы и др.
Исход может быть двояким: устранение вызывающей причины в начале процесса может привести к восстановлению ткани, однако в далеко зашедших случаях наступает гибель клеток.
Значение роговой дистрофии определяется ее степенью, распространенностью и длительностью. Длительно существующее патологическое ороговение слизистой оболочки (лейкоплакия) может явиться источником развития раковой опухоли. Врожденный ихтиоз резкой степени, как правило, несовместим с жизнью.
К группе паренхиматозных диспротеинозов примыкает ряд дистрофий, в основе которых лежат нарушения внутриклеточного метаболизма ряда аминокислот в результате наследственной недостаточности метаболизирующих их ферментов, т.е. в результате наследственной ферментопатии. Эти дистрофии относятся к так называемым болезням накопления.
Наиболее яркими примерами наследственных дистрофий, связанных с нарушением внутриклеточного метаболизма аминокислот, являются цистиноз, тирозиноз,
62
фенилпировиноградная олигофрения (фенилкетонурия). Их характеристика представлена в табл. 1.
Таблица 1. Наследственные дистрофии, связанные с нарушением обмена аминокислот
Название Дефицит фермента
Цистиноз Неизвестен
Тирозиноз
Тирозинаминотрансфераза или оксидаза параоксифенилпировиноградной кислоты
Фенилпирови-
ноградная Фенилаланин-4-гидроксилаза
олигофрения
Паренхиматозные жировые дистрофии (липидозы)
Локализация накоплений амин
Печень, почки, селезенка, глаза, мозг, лимфатические узлы, кожа
Печень, почки, кости
Нервная система, мышцы, кожа, к моча
В цитоплазме клеток содержатся в основном липиды, которые образуют с белками сложные лабильные жиробелковые комплексы - липопротеиды. Эти комплексы составляют основу мембран клетки. Липиды вместе с белками являются составной частью и клеточных ультраструктур. Помимо липопротеидов, в цитоплазме встречаются и нейтральные жиры, которые представляют собой сложные эфиры глицерина и жирных кислот.
Для выявления жиров используют срезы нефиксированных замороженных или фиксированных в формалине тканей. Гистохимически жиры выявляются с помощью ряда методов: судан III и шарлах окрашивают их в красный цвет, судан IV и осмиевая кислота - в черный, сульфат нильского голубого окрашивает жирные кислоты в темно-синий цвет, а нейтральные жиры - в красный.
С помощью поляризационного микроскопа можно дифференцировать изотропные и анизотропные липиды, последние дают характерное двойное лучепреломление.
Нарушения обмена цитоплазматических липидов могут проявляться в увеличении их содержания в клетках, где они обнаруживаются и в норме, в появлении липидов там, где они обычно не встречаются, и в образовании жиров необычного химического состава. Обычно в клетках накапливаются нейтральные жиры.
Паренхиматозная жировая дистрофия встречается наиболее часто там же, где и белковая, - в миокарде, печени, почках.
В миокарде жировая дистрофия характеризуется появлением в мышечных клетках мельчайших жировых капель (пылевидное ожирение). При нарастании изменений эти капли (мелкокапельное ожирение) полностью замещают цитоплазму (рис. 29). Большинство митохондрий при этом распадается, поперечная исчерченность волокон исчезает. Процесс
имеет очаговый характер и наблюдается в группах мышечных клеток, расположенных по ходу венозного колена капилляров и мелких вен.
63
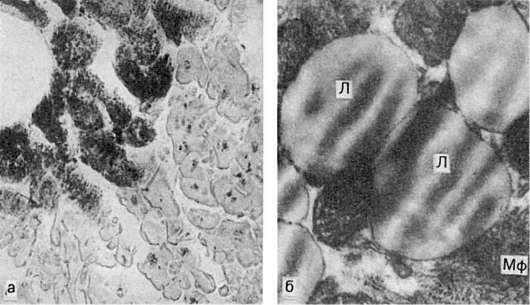
Рис.
а - капли жира (на рисунке черного цвета) в цитоплазме мышечных волокон (микроскопическая картина); б - включения липидов (Л), имеющие характерную исчерченность; Мф - миофибриллы. Электронограмма. х21 000
Внешний вид сердца зависит от степени жировой дистрофии. Если процесс выражен слабо, его можно распознать лишь под микроскопом, применяя специальные окраски на липиды; если он выражен сильно, сердце выглядит увеличенным в объеме, камеры его растянуты, оно дряблой консистенции, миокард на разрезе тусклый, глинисто-желтый. Со стороны эндокарда видна желто-белая исчерченность, особенно хорошо выраженная в сосочковых мышцах и трабекулах желудочков сердца («тигровое сердце»). Эта исчерченность миокарда связана с очаговым характером дистрофии, преимущественным поражением мышечных клеток вокруг венул и вен. Жировая дистрофия миокарда рассматривается как морфологический эквивалент его декомпенсации.
Развитие жировой дистрофии миокарда связывают с тремя механизмами: повышенным поступлением жирных кислот в кардиомиоциты, нарушением обмена жиров в этих клетках и распадом липопротеидных комплексов внутриклеточных структур. Чаще всего эти механизмы реализуются путем инфильтрации и декомпозиции (фанероза) при энергетическом дефиците миокарда, связанном с гипоксией и интоксикацией (дифтерия). При этом основное значение декомпозиции не в высвобождении липидов из липопротеидных комплексов клеточных мембран, а в деструкции митохондрий, что ведет к нарушению окисления жирных кислот в клетке.
В печени жировая дистрофия (ожирение) проявляется резким увеличением содержания жиров в гепатоцитах и изменением их состава. В клетках печени вначале появляются гранулы липидов (пылевидное ожирение), затем мелкие капли их (мелкокапельное ожирение), которые в дальнейшем
64
сливаются в крупные капли (крупнокапельное ожирение) или в одну жировую вакуоль, которая заполняет всю цитоплазму и отодвигает ядро на периферию. Измененные таким образом печеночные клетки напоминают жировые. Чаще отложение жиров в печени начинается на периферии, реже - в центре долек; при значительно выраженной дистрофии ожирение клеток печени имеет диффузный характер.
Внешний вид печени достаточно характерен: она увеличена, дряблая, охряно-желтого или желто-коричневого цвета. При разрезе на лезвии ножа и поверхности разреза виден налет жира.
Среди механизмов развития жировой дистрофии печени различают: чрезмерное поступление в гепатоциты жирных кислот или повышенный их синтез этими клетками; воздействие токсических веществ, блокирующих окисление жирных кислот и синтез липопротеидов в гепатоцитах; недостаточное поступление в печеночные клетки аминокислот, необходимых для синтеза фосфолипидов и липопротеидов. Из этого следует, что жировая дистрофия печени развивается при липопротеидемии (алкоголизм, сахарный диабет, общее ожирение, гормональные расстройства), гепатотропных интоксикациях (этанол, фосфор, хлороформ и др.), нарушениях питания (недостаток белка в пище - алипотропная жировая дистрофия печени, авитаминозы, болезни пищеварительной системы).
В почках при жировой дистрофии жиры появляются в эпителии проксимальных и дистальных канальцев. Обычно это нейтральные жиры, фосфолипиды или холестерин, который обнаруживают не только в эпителии канальцев, но и в строме. Нейтральные жиры в эпителии узкого сегмента и собирательных трубок встречаются как физиологическое явление.
Внешний вид почек: они увеличены, дряблые (при сочетании с амилоидозом плотные), корковое вещество набухшее, серое с желтым крапом, заметным на поверхности и разрезе.
Механизм развития жировой дистрофии почек связан с инфильтрацией эпителия почечных канальцев жиром при липемии и гиперхолестеринемии (нефротический синдром), что ведет к гибели нефроцитов.
Причины жировой дистрофии разнообразны. Чаще всего она связана с кислородным голоданием (тканевая гипоксия), поэтому жировая дистрофия так часто встречается при заболеваниях сердечно-сосудистой системы, хронических заболеваниях легких, анемиях, хроническом алкоголизме и т.д. В условиях гипоксии страдают в первую очередь отделы органа, находящиеся в функциональном напряжении. Вторая причина - инфекции (дифтерия, туберкулез, сепсис) и интоксикации (фосфор, мышьяк, хлороформ), ведущие к нарушениям обмена (диспротеиноз, гипопротеинемия, гиперхолестеринемия), третья - авитаминозы и одностороннее (с недостаточным содержанием белков) питание, сопровождающееся дефицитом ферментов и липотропных факторов, которые необходимы для нормального жирового обмена клетки.
65
Исход жировой дистрофии зависит от ее степени. Если она не сопровождается грубым поломом клеточных структур, то, как правило, оказывается обратимой. Глубокое нарушение обмена клеточных липидов в
большинстве случаев заканчивается гибелью клетки, функция органов при этом резко нарушается, а в ряде случаев и выпадает.
Группу наследственных липидозов составляют так называемые системные липидозы, возникающие вследствие наследственного дефицита ферментов, участвующих в метаболизме определенных липидов. Поэтому системные липидозы относят
к наследственным ферментопатиям (болезни накопления), поскольку дефицит фермента определяет накопление субстрата, т.е. липидов, в клетках.
В зависимости от вида накапливающихся в клетках липидов различают: цереброзидлипидоз, илиглюкозилцерамидлипидоз (болезнь
Гоше), сфингомиелинлипидоз (болезнь Ниманна-Пика),ганглиозидлипидоз (болезнь Тея-
Сакса, или амавротическая идиотия), генерализованный ганглиозидоз(болезнь НорманаЛандинга) и др. Чаще всего липиды накапливаются в печени, селезенке, костном мозге, центральной нервной системе (ЦНС), нервных сплетениях. При этом появляются характерные для того или иного вида липидоза клетки (клетки Гоше, клетки Пика), что имеет диагностическое значение при изучении биоптатов (табл. 2).
Таблица 2. Системные липидозы (наследственные ферментопатии, болезни накопления, лизосомные болезни)
Название |
Дефицит фермента |
Локализация накоплений |
Диагностически |
липида |
критерий при б |
Болезнь Гоше - цереброзидлипидоз или глюкозидцерамидлипидоз Болезнь НиманнаПика - сфингомиелинлипидоз
Амавротическая идиотия, болезнь Тея-Сакса - ганглиозидлипидоз
Болезнь НорманаЛандинга - генерализованный ганглиозидоз
|
Печень, селезенка, костный |
|
|
Глюкоцереброзидазамозг, ЦНС (у детей) |
Клетки Гоше |
||
Сфингомиелиназа |
Печень, селезенка, костный |
Клетки Пика |
|
мозг, ЦНС |
|||
|
|
||
|
ЦНС, сетчатка глаз, |
Изменения |
|
Гексозаминидаза |
нервные сплетения, |
мейсснеровского |
|
|
селезенка, печень |
(ректобиопсия) |
|
|
ЦНС, нервные сплетения, |
|
|
β-Галактозидаза |
печень, селезенка, костный |
Отсутствует |
|
|
мозг, почки и др. |
|
|
Многие ферменты, дефицит которых определяет развитие системных липидозов, относятся, как видно из табл. 2, к лизосомным. На этом основании ряд липидозов рассматривают как лизосомные болезни.
Паренхиматозные углеводные дистрофии
66
Углеводы, которые определяются в клетках и тканях и могут быть идентифицированы гистохимически, делят наполисахариды, из которых в животных тканях выявляются лишь гликоген, гликозаминогликаны (му-
кополисахариды) и гликопротеиды. Среди гликозаминогликанов различают нейтральные, прочно связанные с белками, и кислые, к которым относятся гиалуроновая, хондроитинсерная кислоты и гепарин. Кислые гликозаминогликаны как биополимеры способны вступать в непрочные соединения с рядом метаболитов и осуществлять их транспорт. Главными представителями гликопротеидов являются муцины и мукоиды. Муцины составляют основу слизи, продуцируемой эпителием слизистых оболочек и железами, мукоиды входят в состав многих тканей.
Полисахариды, гликозаминогликаны и гликопротеиды выявляются ШИКреакцией или реакцией Хочкиса-Мак-Мануса. Сущность реакции заключается в том, что после окисления йодной кислотой (или реакции с перйодатом) образующиеся альдегиды дают с фуксином Шиффа красное окрашивание. Для выявления гликогена ШИК-реакцию дополняют ферментативным контролем - обработкой срезов амилазой. Гликоген окрашивается кармином Беста в красный цвет. Гликозаминогликаны и гликопротеиды определяют с помощью ряда методов, из которых наиболее часто применяют окраски толуидиновым синим или метиленовым синим. Эти окраски позволяют выявлять хромотропные вещества, дающие реакцию метахромазии. Обработка срезов ткани гиалуронидазами (бактериальной, тестикулярной) с последующей окраской теми же красителями позволяет дифференцировать различные гликозаминогликаны.
Паренхиматозная углеводная дистрофия может быть связана с нарушением обмена гликогена илигликопротеидов.
Углеводные дистрофии, связанные с нарушением обмена гликогена
Основные запасы гликогена находятся в печени и скелетных мышцах. Гликоген печени и мышц расходуется в зависимости от потребностей организма (лабильный гликоген). Гликоген нервных клеток, проводящей системы сердца, аорты, эндотелия, эпителиальных покровов, слизистой оболочки матки, соединительной ткани, эмбриональных тканей, хряща и лейкоцитов является необходимым компонентом клеток, и его содержание не подвергается заметным колебаниям (стабильный гликоген). Однако деление гликогена на лабильный и стабильный условно.
Регуляция обмена углеводов осуществляется нейроэндокринным путем. Основная роль принадлежит гипоталамической области, гипофизу (АКТГ, тиреотропный, соматотропный гормоны), (β-клеткам (В-клеткам) поджелудочной железы (инсулин), надпочечникам (глюкокортикоиды, адреналин) и щитовидной железе.
Нарушения содержания гликогена проявляются в уменьшении или увеличении количества его в тканях и появлении там, где он обычно не выявляется. Эти нарушения наиболее ярко
67
выражены при сахарном диабете и при наследственных углеводных дистрофиях - гликогенозах.
При сахарном диабете, развитие которого связывают с патологией β-клеток островков поджелудочной железы, происходят недостаточное использование глюкозы тканями, увеличение ее содержания в крови (гипергликемия) и выведение с мочой (глюкозурия). Тканевые запасы гликогена резко уменьшаются. Это в первую очередь касается печени,
в которой нарушается синтез гликогена, что ведет к инфильтрации ее жирами - развивается жировая дистрофия печени; при этом в ядрах гепатоцитов появляются включения гликогена, они становятся светлыми («дырчатые», «пустые», ядра).
С глюкозурией связаны характерные изменения почек при диабете. Они выражаются в гликогенной инфильтрации эпителия канальцев, главным образом узкого и дистального
сегментов. Эпителий становится высоким, со светлой пенистой цитоплазмой; зерна гликогена видны и в просвете канальцев. Эти изменения отражают состояние синтеза гликогена (полимеризация глюкозы) в канальцевом эпителии при резорбции богатого глюкозой ультрафильтрата плазмы.
При диабете страдают не только почечные канальцы, но и клубочки, их капиллярные петли, базальная мембрана которых становится значительно более проницаемой для сахаров и белков плазмы. Возникает одно из проявлений диабетической микроангиопатии -
интеркапиллярный (диабетический) гломерулосклероз.
Наследственные углеводные дистрофии, в основе которых лежат нарушения обмена гликогена, называютсягликогенозами. Гликогенозы обусловлены отсутствием или недостаточностью фермента, участвующего в расщеплении депонированного гликогена, и
относятся поэтому к наследственным ферментопатиям, илиболезням накопления. В
настоящее время хорошо изучены 6 типов гликогенозов, обусловленных наследственной недостаточностью 6 различных ферментов. Это болезни Гирке (I тип), Помпе (II тип), МакАрдля (V тип) и Герса (VI тип), при которых структура накапливаемого в тканях гликогена не нарушена, и болезни Форбса-Кори (III тип) и Андерсена (IV тип), при которых она резко изменена (табл. 3).
Таблица 3. Гликогенозы (наследственные ферментопатии, болезни накопления)
Название болезни |
Дефицит фермента |
Локализация накоплений гл |
Без нарушения структуры гликогена |
|
|
Гирке (I тип) |
Глюкозо-6-фосфатаза |
Печень, почки |
Помпе (II тип) |
Кислая α-клюкозидаза |
Гладкие и скелетные мышцы, |
Мак-Ардля (V тип) |
Система фосфорилаз мышц |
Скелетные мышцы |
Герса (VI тип) |
Фосфорилаза печени |
Печень |
С нарушением структуры гликогена |
|
|
Форбса-Кори, лимитдекстриноз (III тип)Амило-1,6-глюкозидаза |
Печень, мышцы, сердце |
|
Андерсена, амилопектиноз (IV тип) |
Амило-(1,4-1,6)-трансглюкозидазаПечень, селезенка, лимфатич |
|
|
|
68 |
Морфологическая диагностика гликогеноза того или иного типа возможна при биопсии с помощью гистоферментативных методов.
Углеводные дистрофии, связанные с нарушением обмена гликопротеидов
При нарушении обмена гликопротеидов в клетках или в межклеточном веществе происходит накопление муцинов и мукоидов, называемых также слизистыми или слизеподобными веществами. В связи с этим при нарушении обмена гликопротеидов говорят о слизистой дистрофии.
Микроскопическое исследование. Оно позволяет выявить не только усиленное слизеобразование, но и изменения физико-химических свойств слизи. Многие секретирующие клетки погибают и десквамируются, выводные протоки желез обтурируются слизью, что ведет к развитию кист. Нередко в этих случаях присоединяется воспаление. Слизь может закрывать просветы бронхов, следствием чего является возникновение ателектазов и очагов пневмонии.
Иногда в железистых структурах накапливается не истинная слизь, а слизеподобные вещества (псевдомуцины). Эти вещества могут уплотняться и принимать характер коллоида. Тогда говорят о коллоидной дистрофии,которая наблюдается, например, при коллоидном зобе.
Причины слизистой дистрофии разнообразны, но чаще всего это воспаление слизистых оболочек в результате действия различных патогенных раздражителей (см. Катаральное воспаление).
Слизистая дистрофия лежит в основе наследственного системного заболевания, называемого муковисцидозом,для которого характерно изменение качества слизи, выделяемой эпителием слизистых желез: слизь становится густой и вязкой, она плохо выводится, что обусловливает развитие ретенционных кист и склероза (кистозный фиброз). Поражаются экзокринный аппарат поджелудочной железы, железы бронхиального дерева, пищеварительного и мочевого тракта, желчных путей, потовые и слезные железы
(подробнее см.Пренатальная патология).
Исход в значительной мере определяется степенью и длительностью повышенного слизеобразования. В одних случаях регенерация эпителия приводит к полному восстановлению слизистой оболочки, в других - она атрофируется, подвергается склерозу, что, естественно, отражается на функции органа.
Стромально-сосудистые дистрофии
Стромально-сосудистые (мезенхимальные) дистрофии развиваются в результате нарушений обмена в соединительной ткани и выявляются в строме органов и стенках сосудов. Они развиваются на территориигистиона, который, как известно, образован отрезком микроциркуляторного русла с окружающими его элементами соединительной ткани (основное вещество, волокнистые структуры, клетки) и нервными волокнами. Понятными становятся в связи с этим преобладание среди механизмов развития стромально-сосудистых
69
дистрофий нарушений транспортных систем трофики, общность морфогенеза, возможность не только сочетания различных видов дистрофии, но и перехода одного вида в другой.
При нарушениях обмена в соединительной ткани, преимущественно в ее межклеточном веществе, накапливаются продукты метаболизма, которые могут приноситься с кровью и лимфой, быть результатом извращенного синтеза или появляться в результате дезорганизации основного вещества и волокон соединительной ткани.
В зависимости от вида нарушенного обмена мезенхимальные дистрофии делят на белковые (диспротеинозы), жировые (липидозы) и углеводные.
Стромально-сосудистые белковые дистрофии (диспротеинозы)
Среди белков соединительной ткани основное значение имеет коллаген, из макромолекул которого строятся коллагеновые и ретикулярные волокна. Коллаген является неотъемлемой частью базальных мембран (эндотелия, эпителия) и эластических волокон, в состав которых, помимо коллагена, входит эластин. Коллаген синтезируется клетками соединительной ткани, среди которых главную роль играют фибробласты. Кроме коллагена, эти клетки синтезируют гликозаминогликаны основного вещества соединительной ткани, которое содержит также белки и полисахариды плазмы крови.
Волокна соединительной ткани имеют характерную ультраструктуру. Они хорошо выявляются с помощью ряда гистологических методов: коллагеновые - окраской пикрофуксиновой смесью (по ван Гизону), эластические - окраской фукселином или орсеином, ретикулярные - импрегнацией солями серебра (ретикулярные волокна являются аргирофильными).
В соединительной ткани, помимо ее клеток, синтезирующих коллаген и гликозаминогликаны (фибробласт, ретикулярная клетка), а также ряд биологически активных веществ (лаброцит, или тучная клетка), находятся клетки гематогенного происхождения, осуществляющие фагоцитоз (полиморфно-ядерные лейкоциты, гистиоциты, макрофаги) и иммунные реакции (плазмобласты и плазмоциты, лимфоциты, макрофаги).
К стромально-сосудистым диспротеинозам относят мукоидное набухание, фибриноидное набухание (фибриноид), гиалиноз, амилоидоз.
Нередко мукоидное набухание, фибриноидное набухание и гиалиноз являются последовательными стадиямидезорганизации соединительной ткани; в основе этого процесса лежат накопление продуктов плазмы крови в основном веществе в результате повышения тканево-сосудистой проницаемости (плазморрагия), деструкция элементов соединительной ткани и образование белковых (белково-полисахаридных) комплексов. Амилоидоз отличается от этих процессов тем, что в состав образующихся белковополисахаридных комплексов входит не встречающийся обычно фибриллярный белок, синтезируемый клетками - амилоидобластами (схема II).
Схема II. Морфогенез стромально-сосудистых диспротеинозов
70