

Огурцов А.Н. Выравнивание белковых последовательностей. – Харьков. НТУ ХПИ, 2015. – 80 с
..pdfМИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ УКРАИНЫ
НАЦИОНАЛЬНЫЙ ТЕХНИЧЕСКИЙ УНИВЕРСИТЕТ «Харьковский политехнический институт»
А. Н. Огурцов
ВЫРАВНИВАНИЕ БЕЛКОВЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Учебно-методическое пособие по курсам «Биоинформатика и информационная биотехнология» и «Биоинформатика и фармакоинформатика»
для студентов направления подготовки «Биотехнология», в том числе для иностранных студентов
Утверждено редакционно-издательским советом университета, протокол № 1 от 04.06.2014 г.
Х а р ь к о в НТУ «ХПИ»
2 0 1 5
УДК 577.3 ББК 28.071.3
О-39
Рецензенты:
В. В. Давыдов, д-р. мед. наук, проф., зав. лаб. возрастной эндокринологии
иобмена веществ ГП "Институт охраны здоровья детей
иподростков АМН Украины"
В. В. Россихин, д-р мед. наук, профессор кафедры урологии Харьковской медицинской академии последипломного образования МОЗ Украины
Навчально-методичний посібник містить матеріали з основних питань курсів «Біоінформатика та інформаційна біотехнологія» та «Біоінформатика та фармакоінформатика», які є необхідними для самостійного розв'язання задач по вирівнюванню білкових послідовностей.
Призначено для студентів спеціальностей біотехнологічного профілю всіх форм навчання.
Огурцов А. Н.
О 39 Выравнивание белковых последовательностей : учеб.-метод. пособие / А. Н. Огурцов. – Харьков : НТУ «ХПИ», 2015. – 80 с. – На рус. яз.
ISBN 978-617-05-0141-7
Учебно-методическое пособие содержит материалы по основным вопросам курсов «Биоинформатика и информационная биотехнология» и «Биоинформатика и фармакоинформатика», необходимым для самостоятельного решения задач по выравниванию белковых последовательностей.
Предназначено для студентов специальностей биотехнологического профиля всех форм обучения.
Ил. 12. Табл. 8. |
Библиогр.: 12 назв. |
|
УДК 577.3 |
|
ББК 28.071.3 |
ISBN 978-617-05-0141-7 |
А.Н. Огурцов, 2015 |
ВСТУПЛЕНИЕ
Предметом учебных дисциплин "Биоинформатика и информационная биотехнология" и "Биоинформатика и фармакоинформатика" являются компьютерно-ориентированные методы решения информационных задач в области промышленной и фармацевтической биотехнологий. Научную основу курсов составляют молекулярная биофизика, молекулярная биология и общая и молекулярная генетика. Методическими основами курса являются лекции, в которых излагаются основные положения каждого раздела, практические занятия и самостоятельная работа студентов, являющаяся основным способом усвоения материала в свободное от аудиторных занятий время.
Понятие "информация" проникает во все сферы деятельности человека, объединяя их в единый взаимосвязанный и взаимозависимый комплекс. Относительно недавно появился даже термин "инфосфера" – информационные структуры, системы и процессы в науке, обществе и производстве. Вместе с тем до сих пор отсутствует единая точка зрения на предмет информатики, и до сих пор не вполне ясны соотношения между различными информационными дисциплинами, связанными с различными предметными областями.
Интуитивно ясно, что биоинформатика нацелена на использование информации и информационных технологий при исследовании биологических систем. В биоинформатике биология, информатика и
3
математика сливаются в единую дисциплину. В каком-то смысле биоинформатика, изучающая применение информационных технологий для управления биологическими данными, является продолжением вычислительной биологии, изучающей применение методов количественного анализа в моделировании биологических систем.
Интенсивность исследования геномов различных организмов с каждым годом нарастает, ежегодно появляются новые базы данных, в которых хранится информация об исследованных геномах, а уже существующие базы данных непрерывно наращивают свои мощности. Следовательно, с такой же огромной скоростью растёт и объём доступной исследователям биологической информации. Без использования современных информационных технологий уже невозможно ни отыскать, ни обработать ту конкретную биологическую информацию, которая необходима в данном исследовании или в данном биотехнологическом процессе.
Биоинформатика – это наука о хранении, извлечении, организации, анализе, интерпретации и использовании биологической информации. Целью информационной биотехнологии вообще и фармакоинформатики как частного применения информационной биотехнологии для открытия и разработки лекарственных препаратов, является использование существующих и разработка новых компьютерных и информационных ресурсов для анализа и интерпретации биологических данных различного типа (последовательностей ДНК, РНК и белков, пространственных структур РНК и белков, профилей экспрессии, метаболических путей и др.) с целью разработки новых биотехнологических продуктов.
Настоящее пособие подготовлено на основе исправленных и дополненных пособий [1–4] и адаптированных работ [5–11], послуживших также источником иллюстраций, таким образом, чтобы максимально облегчить овладение практическими навыками решения задач, связанных с выравниванием белковых последовательностей, которые затем используются в ходе выполнения индивидуального задания, практических занятий, при подготовке к выполнению контрольных работ и к экзамену.
1. ВЫРАВНИВАНИЕ БИОЛОГИЧЕСКИХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Выравниванием (alignment) последовательностей азотистых осно-
ваний в нуклеиновых кислотах или аминокислот в полипептидных цепях белков называют определение взаимного соответствия остатков (нукле-
иновых оснований или аминокислотных остатков, соответственно) в этих двух или нескольких последовательностях, при котором сохраняется исходный порядок остатков в последовательностях [1]. Одна из целей выравнивания последовательностей состоит в том, чтобы определить степень подобия двух последовательностей и, если она достаточно высока, сделать правдоподобное заключение об их гомологичности.
При передаче генетической информации от предыдущего поколения следующему она несколько изменяется во время процесса копирования. Изменения, которые происходят в процессе расхождения от общего предка, могут быть трёх типов: замены, вставки и удаления (выпадения). Эти изменения могут накапливаться от поколения к поколению. Через несколько тысяч поколений в последовательностях может наблюдаться значительное число расхождений [2]. Сравнение двух предположительно гомологичных последовательностей показывает степень их расхождения, то есть силу эволюционных изменений.
Выравнивание последовательностей – это процедура сравнения двух
(попарное выравнивание) или нескольких (множественное выравнивание)
последовательностей путём поиска рядов отдельных элементов или характерных комбинаций элементов последовательностей, которые расположены в выравниваемых последовательностях в одинаковом порядке.
При выравнивании двух последовательностей их помещают в две строки друг над другом, записывая их с помощью букв алфавита.
Идентичные или подобные "буквы" (элементы) этих строк (последовательностей) сдвигают в пределах строки (не меняя исходного порядка следования "знаков") таким образом, чтобы они выстраивались друг под другом в соответствующих столбцах. Неидентичные, или различные знаки либо помещают в одни и те же столбцы как несовпадения, либо вставляют напротив них во второй последовательности пропуски.
4 |
5 |
Рассмотрим для примера две строки:
1) abcde |
2) acdef |
Разумное выравнивание выглядит так:
abcde- a-cdef
Для того чтобы найти оптимальное (или необходимо определить критерий качества последовательностей нуклеотидов gctgaacg следующие выравнивания:
наилучшее) выравнивание выравнивания. Так, для и ctataatc возможны
1. |
Неинформативное выравнивание |
--------gctgaacg |
|
|
ctataatc-------- |
2. |
Выравнивание без пропусков |
gctgaacg |
|
|
ctataatc |
3. |
Выравнивание с пропусками |
gctga-a--cg |
|
|
--ct-ataatc |
4. |
Ещё одно выравнивание |
gctg-aa-cg |
|
|
-ctataatc- |
Интуитивно кажется, что последнее выравнивание является лучшим, поскольку в нём получено максимальное число совпадений для нуклеотидов в двух последовательностях и использовано минимальное число вставок.
Чтобы решить, является ли оно лучшим из всех возможных, необходимо иметь способ систематической проверки всех возможных выравниваний, иметь количественный критерий ("вес" ("weight") или
счёт ("score")), по которому возможно сравнивать качество различных выравниваний и определить выравнивание с оптимальным весом (счётом).
При этом от того, какая именно система оценки выбрана для такого сравнения, может зависеть результат сравнения, и даже незначительные изменения в схеме оценки могут изменить рейтинг выравниваний, из-за чего лучшим станет другое выравнивание.
Разделяют несколько типов выравнивания.
6
Глобальное выравнивание – это выравнивание всей последовательности относительно другой последовательности.
Я.люблю.тебя,.жизнь,.Что.само-.по.себе-.и.не.ново.
||||||||||||||||||||| || ||| ||| |
Я.люблю.тебя,.жизнь,.я.люблю.тебя.снова.и.--снова.
Здесь символом "|" обозначены соответствия, "пробелы" обозначают несоответствия, "-" обозначает те вставки (инсерции, от англ. insertion) и удаления (делеции, от англ. deletion), которые необходимо сделать в обеих последовательностях, чтобы достичь максимального количества соответствий.
Локальное выравнивание – это поиск части последовательности, которая совпадает с частью другой последовательности.
Ямщик, не гони лошадей! Мне некуда больше спешить,
|||||||||||||||||||||||
Мне некого больше любить, Ямщик, не гони лошадей!
Для локального совпадения выступающие концы не рассматриваются как пропуски (делеции). В дополнение к несовпадениям, возможны также вставки и удаления внутри совпадающей части.
Поиск мотивов совпадения – это поиск совпадения короткой последовательности в одном или более отрезках длинной последовательности. В этом случае допускается несовпадение одного символа. Можно также потребовать полного совпадения, либо допустить бόльшее число несовпадений или даже пропусков.
Для примера, найдём мотивы «Я люблю» совпадения для строк
Ялюблю
|||||||
Ялюблю тебя, жизнь, Что само по себе и не ново.
Я люблю |
я |
люблю |
||||||| |
||||||| |
|
Я люблю тебя, жизнь, я |
люблю тебя снова и снова. |
|
Вот уж окна зажглись, Я шагаю с работы устало, -
Ялюблю
|||||||
Ялюблю тебя, жизнь, И хочу, чтобы лучше ты стала.
7
Множественное выравнивание – это взаимное выравнивание многих последовательностей. Например, выровняем четыре строки:
Никто,.никто,.-.сказал.он.И.вылез.-------из.кровати-, Никто,.никто,.-.сказал.он,.Спускаясь.--вниз.в.халате, Никто,.никто,.-.сказал.он,.Намылив.---руки.мылом----, Никто,.никто,.-.сказал.он,.Съезжая.по.перилам-------,
Никто,.никто,.-.сказал.он----------------и----------,
Последняя, пятая строка, показывающая символы, сохраненные во всех последовательностях выравнивания, называется консенсусом.
2. ТОЧЕЧНАЯ МАТРИЦА СХОДСТВА
Точечная матрица (dot plot) представляет собой таблицу или матрицу, в которой строки соответствуют элементам одной последовательности, а колонки – элементам другой последовательности. В простейшем варианте ячейки точечной матрицы оставляют пустыми, если сравниваемые элементы различны, и заполняются, если они совпадают [3]. Для примера построим точечную матрицу, показывающую совпадения между короткой строкой ПРОФЕССОРОГУРЦОВ и длинной
ПРОФЕССОРАЛЕКСАНДРНИКОЛАЕВИЧОГУРЦОВ (рисунок 1).
|
П Р О Ф Е С С О Р А Л Е К С А Н Д Р Н И К О Л А Е В И Ч О Г У Р Ц О В |
||||||||||||||||||||||||||||||||||
П |
П |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Р |
|
Р |
|
|
|
|
|
|
Р |
|
|
|
|
|
|
|
|
Р |
|
|
|
|
|
|
|
|
|
|
|
|
|
Р |
|
|
|
О |
|
|
О |
|
|
|
|
О |
|
|
|
|
|
|
|
|
|
|
|
|
|
О |
|
|
|
|
|
|
О |
|
|
|
|
О |
|
Ф |
|
|
|
Ф |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Е |
|
|
|
|
Е |
|
|
|
|
|
|
Е |
|
|
|
|
|
|
|
|
|
|
|
|
Е |
|
|
|
|
|
|
|
|
|
|
С |
|
|
|
|
|
С |
С |
|
|
|
|
|
|
С |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
С |
|
|
|
|
|
С |
С |
|
|
|
|
|
|
С |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
О |
|
|
О |
|
|
|
|
О |
|
|
|
|
|
|
|
|
|
|
|
|
|
О |
|
|
|
|
|
|
О |
|
|
|
|
О |
|
Р |
|
Р |
|
|
|
|
|
|
Р |
|
|
|
|
|
|
|
|
Р |
|
|
|
|
|
|
|
|
|
|
|
|
|
Р |
|
|
|
О |
|
|
О |
|
|
|
|
О |
|
|
|
|
|
|
|
|
|
|
|
|
|
О |
|
|
|
|
|
|
О |
|
|
|
|
О |
|
Г |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Г |
|
|
|
|
|
У |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
У |
|
|
|
|
Р |
|
Р |
|
|
|
|
|
|
Р |
|
|
|
|
|
|
|
|
Р |
|
|
|
|
|
|
|
|
|
|
|
|
|
Р |
|
|
|
Ц |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ц |
|
|
О |
|
|
О |
|
|
|
|
О |
|
|
|
|
|
|
|
|
|
|
|
|
|
О |
|
|
|
|
|
|
О |
|
|
|
|
О |
|
В |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
В |
|
|
|
|
|
|
|
|
В |
Рисунок 1 – Точечная матрица сходства двух строк
Буквы, соответствующие длинным совпадающим участкам, выделены жирным шрифтом, а одиночные совпадения, не выделены жирным шрифтом. Очевидно, выравнивание в этом случае будет иметь вид
ПРОФЕССОРАЛЕКСАНДРНИКОЛАЕВИЧОГУРЦОВ
ПРОФЕССОР------------------- |
ОГУРЦОВ |
На рисунке 2 представлена точечная матрица, показывающая как |
|
глобальные, так и локальные совпадения повторяющейся последовательности АБРАКАДАБРАКАДАБРА с самой собой.
А Б Р А К А Д А Б Р А К А Д А Б Р А
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
Б |
|
Б |
|
|
|
|
|
|
Б |
|
|
|
|
|
|
Б |
|
|
Р |
|
|
Р |
|
|
|
|
|
|
Р |
|
|
|
|
|
|
Р |
|
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
К |
|
|
|
|
К |
|
|
|
|
|
|
К |
|
|
|
|
|
|
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
Д |
|
|
|
|
|
|
Д |
|
|
|
|
|
|
Д |
|
|
|
|
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
Б |
|
Б |
|
|
|
|
|
|
Б |
|
|
|
|
|
|
Б |
|
|
Р |
|
|
Р |
|
|
|
|
|
|
Р |
|
|
|
|
|
|
Р |
|
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
К |
|
|
|
|
К |
|
|
|
|
|
|
К |
|
|
|
|
|
|
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
Д |
|
|
|
|
|
|
Д |
|
|
|
|
|
|
Д |
|
|
|
|
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
Б |
|
Б |
|
|
|
|
|
|
Б |
|
|
|
|
|
|
Б |
|
|
Р |
|
|
Р |
|
|
|
|
|
|
Р |
|
|
|
|
|
|
Р |
|
А |
А |
|
|
А |
|
А |
|
А |
|
|
А |
|
А |
|
А |
|
|
А |
Рисунок 2 – Точечная матрица совпадений в повторяющейся последователь-
ности
Вид точечной матрицы может наглядно показать наличие палиндромных последовательностей в анализируемой строке.
Длинные участки ДНК или РНК, содержащие инвертированные повторы такого типа, могут формировать шпилечные структуры. Кроме того, некоторые подвижные элементы, выделенные из растений, содержат
8 |
9 |

настоящие (неточные) палиндромные последовательности – инвертированные повторы некомплементарных последовательностей, расположенных на той же цепи.
На рисунке 3 показан характерный вид точечной матрицы палин-
дрома АРОЗАУПАЛАНАЛАПУАЗОРА.
А Р О З А У П А Л А Н А Л А П У А З О Р А
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
|
|
|
А |
|
|
|
А |
Р |
|
Р |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Р |
|
О |
|
|
О |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
О |
|
|
З |
|
|
|
З |
|
|
|
|
|
|
|
|
|
|
|
|
|
З |
|
|
|
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
А |
|
|
А |
|
|
|
А |
У |
|
|
|
|
|
У |
|
|
|
|
|
|
|
|
|
У |
|
|
|
|
|
П |
|
|
|
|
|
|
П |
|
|
|
|
|
|
|
П |
|
|
|
|
|
|
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
А |
|
|
А |
|
|
|
А |
Л |
|
|
|
|
|
|
|
|
Л |
|
|
|
Л |
|
|
|
|
|
|
|
|
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
А |
|
|
А |
|
|
|
А |
Н |
|
|
|
|
|
|
|
|
|
|
Н |
|
|
|
|
|
|
|
|
|
|
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
А |
|
|
А |
|
|
|
А |
Л |
|
|
|
|
|
|
|
|
Л |
|
|
|
Л |
|
|
|
|
|
|
|
|
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
А |
|
|
А |
|
|
|
А |
П |
|
|
|
|
|
|
П |
|
|
|
|
|
|
|
П |
|
|
|
|
|
|
У |
|
|
|
|
|
У |
|
|
|
|
|
|
|
|
|
У |
|
|
|
|
|
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
А |
|
|
А |
|
|
|
А |
З |
|
|
|
З |
|
|
|
|
|
|
|
|
|
|
|
|
|
З |
|
|
|
О |
|
|
О |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
О |
|
|
Р |
|
Р |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Р |
|
А |
А |
|
|
|
А |
|
|
А |
|
А |
|
А |
|
А |
|
|
А |
|
|
|
А |
Рисунок 3 – Точечная матрица сходства (совпадений) для палиндромной последовательности АРОЗАУПАЛАНАЛАПУАЗОРА
Фрагмент генома вируса Wheat Dwarf Virus, вызывающего остановку роста пшеницы – ttttcgtgagtgcggaggctttt – это пример нуклеотидного палиндрома.
Точечная матрица позволяет быстро проиллюстрировать родство между двумя последовательностями. Яркие признаки сходства чётко проявляются. Например, точечная матрица, отображающая родство
10
между генами митохондриальной АТФазы миноги Petromyzon marinis
(lamprey) и морской собаки Scyliorbinus canicula (dogfish), показывает, что сходство между этими последовательностями менее всего выражено вначале (рисунок 4).
Рисунок 4 – Точечная матрица совпадений для АТФазы-6 из миноги и морской
собаки
Точечная матрица не просто визуализирует сходство двух последовательностей, она вообще демонстрирует все возможные выравнивания и отображает их относительное качество.
Выравнивание не должно изменять "смысл" последовательностей, поэтому при выравнивании должна сохраняться последовательность символов в строке и не должно быть перестановок символов. Поэтому при построении выравнивания, начиная с верхнего левого угла точечной матрицы, разрешены только три типа шагов:
1)строго направо ();
2)строго вниз ();
3)по диагонали слева направо и сверху вниз ().
11
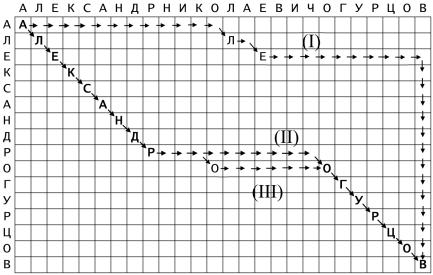
Любой путь по точечной матрице от левого верхнего угла к правому нижнему углу, построенный с помощью этих шагов, соответствует одному из возможных выравниваний.
Например, на рисунке 5 приведены три варианта выравнивания строк АЛЕКСАНДРНИКОЛАЕВИЧОГУРЦОВ и АЛЕКСАНДРОГУРЦОВ:
(I)
АЛЕКСАНДРНИКОЛАЕВИЧОГУРЦОВ–––––––––––––
А---- |
–------- |
Л-Е---------- |
КСАНДРОГУРЦОВ |
(II) |
|
|
|
|
|
АЛЕКСАНДРНИКОЛАЕВИЧОГУРЦОВ |
|
|
|
АЛЕКСАНДР---------- |
ОГУРЦОВ |
(III) |
|
|
|
|
|
АЛЕКСАНДРНИКОЛАЕВИЧОГУРЦОВ |
|
|
|
АЛЕКСАНДР---О------- |
ГУРЦОВ |
Рисунок 5 – Возможные варианты выравнивания
Любой путь по точечной матрице от верхнего левого угла к нижнему правому проходит последовательность ячеек, каждая из которых предсказывает пару позиций: одну из ряда и одну из столбца, которые совпадают с выравниванием; либо означают пробел в одной из последовательностей. Путь не обязательно должен проходить лишь заполненные позиции. Тем не менее, чем больше заполненных позиций, на диагональном отрезке пути, тем больше совпадающих остатков в выравнивании.
Если направление движения между последующими ячейками диагональное (), то пара следующих друг за другом сравниваемых остатков оказываются в выравнивании без вставки между ними
(сопоставляются).
Если направление движения горизонтальное (), то в последовательность, служащую указателем рядов, вставляется пропуск.
Если же направление движения вертикальное (), то пропуск вставляется в последовательность, индексирующую столбцы.
Следует обратить внимание на то, что ни одно движение не может совершаться вверх или влево, так как это соответствовало бы сравнению нескольких остатков одной последовательности со всего лишь одним остатком другой. Математическая интерпретация изложенного выше способа выбора пути по точечной матрице основывается на представлении пути выравнивания в виде графа.
Граф определяется как совокупность множества вершин (или узлов) и множества связей между узлами, которые называются рёбра (или дуги).
Ориентированный граф (кратко орграф) – это (мульти) граф,
рёбрам которого присвоено направление.
Маршрутом в орграфе называют чередующуюся последовательность вершин и дуг (вершины могут повторяться). Длина маршрута – это количество дуг в нем.
Путь – это маршрут в орграфе без повторяющихся дуг; простой путь – без повторяющихся вершин. Если существует путь из одной вершины в другую, то вторая вершина достижима из первой.
12 |
13 |
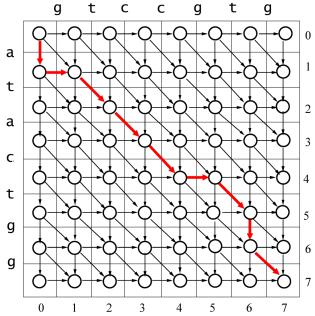
Рассмотрим две последовательности длиной m и n . Выравниванием этих последовательностей будет ориентированный граф G с узлами (i, j) ( 0 i m, 0 j n ) решётки размером (m 1) (n 1) . Ребро графа от узла (i, j) к узлу (i , j ) возможно только если 0 i i 1 и 0 j j 1.
На рисунке 6 представлен граф выравнивания для последовательностей X = gtccgtg и Y = atactgg, в котором существуют вертикальные, горизонтальные и диагональные рёбра.
Рисунок 6 – Граф выравнивания последовательностей gtccgtg и atactgg
В ориентированном графе концом графа (или стόком, sink) является узел, к которому направлены все прилегающие рёбра, а началом графа (или источником, source) является узел, от которого направлены все прилегающие рёбра. В графе глобального выравнивания единственным началом (source) является узел (0,0) , а единственным концом графа (sink) выравнивания является узел (m, n) .
14
Выделенный жирными стрелками путь на графе выравнивания (рисунок 6) можно представить в виде
s (0,1)(1,1)(2,2)(3,3)(4,4)(5,4)(6,5)(6,6)(7,7),
где s – начало (0,0). Соответствующее выравнивание будет иметь вид
X-gtccgt-g
Ya-tac-tgg
Другой способ интерпретации пути по точечной матрице – это порядок редактирования (edit script). Он представляет собой указание серий операций, которые трансформируют последовательность, индексирующую столбцы, – горизонтально расположенную последовательность (над таблицей) – в последовательность, которая индексирует ряды, – вертикально расположенную последовательность (слева от таблицы).
Каждое движение говорит нам о проведении одной из операций –
замены, вставки или делеции (удаления). По достижении конца пути получится преобразование одной последовательности в другую.
Вообще говоря, несколько различных последовательностей редакционных операций могут преобразовать одну последовательность в другую за одно и то же количество шагов, при этом, однако, они могут соответствовать различным выравниваниям.
Оптимальным называют выравнивание с максимальным счётом, наибольшим числом соответствий и наименьшим количеством различий.
Субоптимальное выравнивание – это условно оптимальное выравнивание, где наивысший счёт находится ниже оптимального уровня.
При оптимальном выравнивании неидентичные знаки и пропуски размещают так, чтобы в столбцах выравнивания было как можно больше идентичных, или подобных знаков.
Оптимальные выравнивания позволяют выявлять эволюционные отношения последовательностей, предоставляя возможно лучшую информацию относительно того, какие знаки последовательностей должны стоять в одних и тех же столбцах выравнивания, а какие являются вставками в одной из последовательностей (или, соответственно,
15
выпадениями в другой). Эта информация необходима для предсказания функций, структур и эволюционных отношений последовательностей по их выравниванию.
Для того чтобы количественно сравнивать результаты различных вариантов выравнивания, операциям редактирования сопоставляется определённая схема оценок.
3. ВЕС ОПЕРАЦИЙ РЕДАКТИРОВАНИЯ
Не все замены элементов биологических последовательностей равноценны. Так, например, консервативные замены аминокислот сохраняют функциональность белка, а радикальные могут привести к потере функции. Кроме того, высокая помехоустойчивость генетического кода приводит к тому, что консервативные мутации происходят с бόльшей вероятностью, чем радикальные (см. [7], п. 4.2.1). Другой пример, делеция расположенных рядом нуклеиновых оснований в молекулах нуклеиновых кислот или расположенных рядом аминокислот в белках – это событие более вероятное, чем делеция такого же количества позиций, независимо расположенных в биологической последовательности. Поэтому при расчёте расстояния между последовательностями каждому виду редактирования назначается различный "вес" (weight) или "цена" ("счёт") (score).
Чтобы получить оптимальный вес необходимо добавлять баллы за каждую пару совпадающих знаков в выравнивании (диагональные переходы на рисунке 6) и вычитать баллы (штрафовать) за вставки и удаления или делеции (вертикальные и горизонтальные переходы на рисунке 6).
В различных задачах применяют три вида штрафов за делеции:
1)фиксированный штраф: d ;
2)линейный штраф, при котором цена удаления g остатков определяется линейной функцией: (g) gd ;
3) аффинный |
штраф: (g) d (g 1)e , складывающийся |
из |
штрафа d |
за введение делеции (gap-open) и штрафа e |
за |
продолжение делеции (gap-extension). |
|
|
4. МАТРИЦЫ ЗАМЕН АМИНОКИСЛОТ
Для аминокислотных последовательностей было предложено несколько схем замен аминокислот. Известно, что некоторые виды замен аминокислот обычно наблюдаются в родственных белках у организмов разных видов. Поскольку белок с этими заменами остается функционально активным, то, очевидно, что замещающие аминокислоты совместимы с его структурой и функцией. Часто такие замены происходят между химически подобными аминокислотами, однако появляются также изменения другого вида, хотя относительно редко.
Знание частот появления замещений всех типов, происходящих в различных белках (из большой выборки) может помочь в предсказании выравниваний любого набора белковых последовательностей. Если последовательности родственных белков вполне подобны, то их легко выровнять и можно без труда отследить все замены аминокислот, наступившие на последней стадии эволюции. Если наследственные отношения среди группы белков предварительно установлены, то можно предсказать наиболее вероятные замены аминокислот, произошедшие в ходе эволюции.
Данный метод анализа был предложен и внедрен в научную практику Маргарет Дейхофф. Она собирала статистику по частотам аминокислотных замен в известных белках, и результаты её работ использовались для подсчёта весов выравниваний в течение многих лет. Позднее они были заменены новыми матрицами, полученными в результате обработки расшифрованных последовательностей.
Матрица PAM (Percent Accepted Mutation) отражает вероятность замен аминокислот в ходе эволюционного изменения аминокислотных последовательностей в белковых цепях. Матрицы PAM (п. 9) показывают изменения, ожидаемые в течение определённого периода эволюционного времени и сопровождаемые убывающим подобием последовательностей, по мере того как гены, кодирующие один и тот же белок, расходятся при увеличении времени эволюции.
Мера расхождения последовательностей оценивается в единицах PAM – процент принятых (или зафиксированных) мутаций. Таким
16 |
17 |