

Предмет і завдання біохімії. Основні напрями та розділи біохімії: статична, динамічна, функціональна біохімія, медична та клінічна біохімія. Біологічна хімія - наука, що вивчає склад і структуру хімічних речовин живої матерії та їх перетворення, які лежать в основі життєдіяльності. В.І. Вернадський, перший президент АН України, вважав, що біохімія — це наука про структуру і поведінку живої речовини. Під живою речовиною він розумів сукупність усіх живих організмів, здатних захоплювати з навколишнього середовища певні хімічні елементи і складні речовини, видозмінювати їх і виділяти кінцеві продукти перетворення в навколишнє середовище. Статична біохімія вивчає склад і хімічну структуру тканин та органів. Це найнижчий ступінь пізнання живого на молекулярному рівні. Біохімія приділяє основну увагу значенню певних біомолекул в утворенні клітинних та тканинних структур, реалізації фізіологічних функцій організму. Динамічна біохімія вивчає хімічні реакції, що складають в сукупності обмін речовин, або метаболізм живих організмів. Своїм завданням динамічна біохімія має вивчення перебігу та механізмів реакцій обміну вуглеводів, білків, ліпідів, нуклеїнових кислот. Функціональна біохімія на підставі даних статичної і динамічної біохімії вивчає біохімічні процеси, які лежать в основі функціональної діяльності різних органів і систем. медична біохімія, яка вивчає закономірності обміну речовин та їх порушення в умовах як нормального функціонування людського організму, так і виникнення патологічних процесів. Біохімічні підходи все ширше використовують для розкриття сутності різних видів патології.Підрозділом медичної біохімії є клінічна біохімія, що вивчає біохімічні процеси, які відбуваються в організмі хворої людини, при певних захворюваннях, дослідження яких може бути використаним у діагностиці ураження певних органів, тканин, клітинних структур.
Біохімія як фундаментальна медико-біологічна наука. Історія розвитку, наукові біохімічні школи, значення в системі вищої медичної освіти. Біохімія, як наука, сформувалась на межі ХІХ-ХХ століття. Термін „біохімія” має у своїй основі два грецькі слова: біос і хем. „Біос” означає життя, слово „хем” бере свій початок від назви провінції в гирлі Нілу, що біля Александрії в Єгипті. Тут на врожайних землях здавна оселялися люди і розвивали різні види діяльності. У них було розвинуто виробництво хліба, вина, оцту, дублення шкіри, виготовлення барвників, тобто вони користувалися біохімічними технологіями. Араби, завоювавши в першому тисячолітті Єгипет, не знищили науки, що розвивалася тут, а, навпаки, доповнили та видозмінили їх іншими напрямками. Саме завдяки арабському префіксу „ал” з’явилися такі слова, як алхімія, алгебра, алгоритм тощо. Алхіміки прагнули перетворити прості метали на золото і срібло за допомогою фантастичного „філософського каменя”. Ідеї алхімії зародилися у Китаї, Єгипті, Індії. Вони були дуже поширені в Західній Європі в ХП-ХV ст. Найвидатнішими алхіміками були Р. Бекон, Ф.А.Парацельс (XVI ст.), Йоган Фрідріх Гельвеціус (XVII ст.). Алхіміки нагромадили великий експериментальний матеріал, відкрили ряд хімічних елементів, описали хімічні властивості та реакції, що перебігають із цими елементами, багато працювали з біологічними рідинами. Так, у 1600 р. один з алхіміків, досліджуючи сечу (кип’ятив її з метою одержання золота), помітив, що осад сечі в темряві світиться. Так було відкрито фосфор - елемент життя і мислення, який потрібний усім живим істотам. Алхімік і лікар Ф.А. Парацельс (XVI ст.) вперше заклав основи ятрохімії (медичної хімії). Назва науки походить від грецького слова „ятрос”, що означає лікар. Він вважав, що хімія повинна служити здоров’ю. Ф.А. Парацельс вперше почав лікувати мінеральними водами, запропонував компреси, препарати ртуті й сірки для лікування венеричних захворювань, які в середні віки були дуже поширеними. Інший ятрохімік XVII ст. Ян Батіст Ван-Гельмонт вперше висунув думку, що в „соках” живого тіла є особливі речовини - „ферменти”, які беруть участь у різних хімічних перетвореннях. У Київській Русі внучка Володимира Мономаха Євпраксія Мстиславівна випустила трактат „Мазі” - своєрідну енциклопедію лікарських знань про хімічні речовини, лікарські трави та процедури для лікування хворих. Наш співвітчизник, виходець із Харківщини, О.Я. Данилевський, досліджуючи будову білків, сформулював положення про їх структуру. Ще один український учений - академік І.Я. Горбачевський - вперше виділив амінокислоти з білків і висунув думку про те, що вони є будівельним матеріалом для останніх, синтезував сечову кислоту, відкрив фермент ксантиноксидазу. У цей період було відкрито фосфор - елемент, який потрібний усім живим істотам. Однак найбільшого розвитку біохімія зазнала в XX ст., коли вчені відкрили ряд речовин, біохімічних процесів та механізми їх регуляції. Зокрема, було запропоновано теорію будови білків, розроблено методи синтезу пептидів. У 1902 р. Е. Фішер здійснив штучний синтез пептидів; одержано білок у кристалічному стані (Дж. Самнер і Дж. Нортроп), у 1861 р О. Бутлеров здійснив синтез (поза організмом) вуглеводів із формальдегіду; вивчено основні шляхи перетворення в організмі білків, вуглеводів, ліпідів, окиснення і синтезу жирних кислот та інших ліпідів (Ф. Кнооп, Феодор Фелікс Конрад Лінен, Ф. Ліпман, Мері Рамфо Кеннеді, А. Ленінджер). У цей час створюються схеми шляхів перетворення вуглеводів і утворення при цьому носія енергії — АТФ (Г. Ембден, О.Ф. Мейєргоф, Ф. Діккенс, Г. Кребс, О. Варбург). Важливу роль в обґрунтуванні механізмів перетворення вуглеводів зіграли роботи біохіміків В. Енгельгардта, Я. Парнаса, Л. Іванова
Внесок вчених кафедри біохімії Львівського національного медичного університету імені Данила Галицького в розвиток біологічної хімії. Понад 100 років пройшло з часу створення кафедри біологічної хімії Львівського національного медичного університету імені Данила Галицького.З 1922 по 1941 рік кафедрою біохімії Львівського медичного інституту завідував всесвітньо відомий вчений академік АН СРСР Якуб Оскарович Парнас. Заслуги Я. Парнаса полягають в розробці проблеми анаеробного розпаду вуглеводів, які загальновизнані та позначені в біохімічній літературі як теорія Ембдена-Мейергофа-Парнаса або „схема гліколізу ЕМП”. Я. Парнас один з перших у світі використав радіоізотопний метод в біохімічних дослідженнях. Надзвичайне значення мало встановлення Я. Парнасом того факту, що більш високофосфорильовані похідні аденілової кислоти, у вигляді яких вона знаходиться в м’язових волокнах (і в еритроцитах), а саме АТФ і АДФ, на відміну від самої аденілової кислоти не підлягають в клітинах ферментативному дезамінуванню. Спостереження за процесом амоніогенезу в м’язовій тканині при різних її станах, кількісне визначення взаємоперетворень аденілової кислоти та її похідних в м’язовій тканині в фізіологічних умовах й при різних експериментальних впливах, зв’язку цих перетворень з актом м’язового скорочення, спряження їх з проміжними реакціями глікогенолізу і перетвореннями креатинфосфату в м’язах. Я. Парнасом і його учнями були вивчені біологічні та хімічні властивості м’язової аденілової кислоти та її похідних, був запропонований біологічний метод їх кількісного визначення. Я. Парнас був організатором і довший час науковим керівником хіміко-фармацевтичного інституту Львівського університету, а також діючого і зараз Львівського фармацевтичного заводу. На цьому підприємстві під керівництвом Я. Парнаса та його учня і найближчого співробітника, талановитого професора П. Остерна, було організовано виробництво багатьох важливих біохімічних препаратів для лікувального використання і науково-дослідницької роботи: аденозинтрифосфатної та аденілової кислот, гексозофосфатів, жовчних кислот, гістидину тощо. Я. Парнас був деканом медичного та хіміко-фармацевтичного факультетів, директором Львівського медичного інституту. У Львові він створив великий колектив молодих талановитих учнів, у результаті дослідницької роботи яких, Львівський інститут медичної хімії займав одне з перших місць серед провідних світових центрів біохімії. Колективом було видано понад 300 наукових праць. Я. Парнас є автором близько 170 наукових робіт, в тому числі 5 підручників. У даний час на кафедрі проводяться роботи по вивченню механізмів регуляції органів травної системи, вивчається вплив стресу на цитопротективні та ульцерогенні механізми слизової оболонки органів травлення, метаболічні та іонно-транспортні процеси, досліджується вплив факторів оточуючого середовища на організм, розглядаються питання ендоекології, проводиться розробка нових методів діагностики та лікування. Колектив кафедри є автором низки посібників і підручників з біологічної хімії.
Хімічний склад живого організму. Біомолекули (білки, вуглеводи, ліпіди, нуклеїнові кислоти, гормони, вітаміни тощо), їх біохімічні функції. Характерні риси живої матерії: обмін речовин й енергії та їх зв’язок із зовнішнім середовищем. Біомолекули — біоорганічні сполуки, що входять до складуживих організмів та спеціалізовані для утворення клітинних структур і участі в біохімічних реакціях, які становлять сутність обміну речовин та фізіологічних функцій живих клітин. Функції біомолекул у живих організмах: а) участь у біохімічних реакціях обміну речовин в ролі субстратів та проміжних продуктів (метаболітів). Прикладами є моносахариди та їх фосфорні ефіри, жирні кислоти та продукти їх окислення, амінокислоти, кетокислоти, дикарбонові кислоти, пуринові та піримідинові основи тощо; б) участь в утворенні інших, більш складних молекул — білків, нуклеїнових кислот, полісахаридів, ліпідів (наприклад, амінокислоти, нуклеотиди, вищіжирні кислоти тощо), або біологічних структур (мембран, рибосом, ядерного хрома- тину тощо); в) участь у регуляції біохімічних процесів та фізіологічних функцій окремих клітин та цілісного організму. Біомолекулами-регуляторами є вітаміни, гормони та гормоноподібні сполуки, внутрішньоклітинні регулятори — циклічні нуклеотиди цАМФ, цГМФ тощо. Головні класи біомолекул, що складають основу структури та функції живих організмів. Білки та амінокислоти. Білки (протеїни) — найважливішийкласбіомолекул, знаявністюяких, атакож нуклеїнових кислот, пов’язують саму хімічну сутність життя в умовах Землі. Нуклеїнові кислоти та нуклеотиди. Нуклеїнові кислоти — дезоксирибонуклеїнові (ДНК) та рибонук- леїнові (РНК) — біополімери (біомакромолекули), що складаються з п’яти основних нуклеотидів пуринового та піримідинового ряду, є носіями генетичної інформації у всіх живих організмах, починаючивіднайпростішихвірусівдоорганізмулюдиниЛіпіди та їх похідні — біомолекули різноманітної хімічної будови, головною особливістю яких є їх гідрофобний характер. Ліпіди виконують численні біологічні функції, виступаючиякенергетичнийматеріал (триацилгліцероли, абонейтральніжири), основа структурибіомембран (фосфоліпіди, гліколіпіди), фізіологічноактивні сполуки зрегуляторноюдією (стероїдні гормони, жиророзчинні вітаміни, ейкозаноїди). Вітаміни — сполуки, що не синтезуються в тваринних організмах, але необхідні для життєдіяльності, зокрема є компонентами метаболізму, за участю яких функ- ціонуютьпевнінайважливішіферментнісистеми. Вітаміниповинніпостійнонадходити в організм у складі продуктів харчування, переважно рослинного (більшість водорозчинних вітамінів) аботваринного (деякіжиророзчинні вітаміни) походження. Гормони — біомолекули, що є передавачами хімічних сигналів у системі ендокринноїрегуляції. Завдякирегуляторнійдії гормонів, медіаторівнервовоїсистеми танаявностілокалізованихнаклітинах-мішеняхбіохімічнихструктур, що специфічнимчином реагують на дію цих біорегуляторів зміною своєї функціональної активності (клітиннихрецепторів), відбуваєтьсяінтеграціяокремиханатомо-фізіологічнихсистем уціліснийбагатоклітиннийорганізм.
Структурні елементи прокаріотичних та еукаріотичних клітин. Основні функції субклітинних органел, їх фракційне розділення методом ультрацентрифугування. Незважаючи на багатоманітність форм, організація клітин усіх живих організмів підпорядкована єдиним закономірностям.Так, усі клітини складаються з поверхневого апарату та цитоплазми. Залеж но від наявності ядра всі організми поділяють на два надцарства: Прокаріоти та Еукаріоти. Клітини прокаріотів, крім того, що не мають ядра, ще й досить просто організовані. Клітини еукаріотів – грибів, рослин і тварин – організовані складніше і обов’язково мають ядро. Клітини еукаріотів, хоча й організовані подібно, також мають певні відмінності. Внутрішній вміст кожної клітини оточує поверхневий апарат. До його складу входять плазматична мембрана, надмембранні та підмембранні структури. Поверхневий апарат клітини захищає її внутрішній вміст від несприятливих впливів довкілля, забезпечує обмін речовинами та енергією між клітиною і середовищем, що її оточує. Внутрішнє середовище клітини – це цитоплазма (від грец. китос –клітина та плазма – виліплене). До її складу входять різні органічні та неорганічні сполуки, а також клітинні компоненти: органели та включення. Цитоплазма за допомогою внутрішньоклітинних мембран поділена на окремі функціональні ділянки. У цитоплазмі розташований внутрішньоклітинний скелет, або цитоскелет. Це система білкових утворів – мікротрубочок і мікрониток, яка виконує насамперед опорну функцію. Крім того, елементи цитоскелета беруть участь у зміні форми та русі клітини, забезпечують певне розташування і переміщення органел. Органели (від грец. органон – орган, інструмент) – постійні клітинні структури. Кожна з органел забезпечує відповідні процеси життєдіяль ності клітини (живлення, рух, синтез певних сполук, зберігання і передачу спадкової інформації тощо). Одномембранні органели це вакуолі, комплекс Гольджі, ендоплазматична сітка, лізосоми. Двомембранні органели: хлоропласти, мітохондрії, ядро. Немембранні органели: клітинний центр, рибосоми, мікротрубочки, мікронитки. Особливості будови тієї чи іншої органели тісно пов’язані з її функціями. На відміну від органел, клітинні включення – не постійні компоненти клітини.
Принципи основних методів біохімічних досліджень: Центрифугування. Цей метод полягає в розділенні неоднорідних систем (суспензій, емульсій) в полі дії доцентрових сил. Під дією цих сил суспензії розділяються на тверду фазу – осад і рідку – центрифугат, який ще називають супернатантом або надосадовою рідиною. Розрізняють препаративне й аналітичне центрифугування. Оптичні методи дослідження. Ці методи належать до найпоширеніших у біохімії. Вони дають можливість одержати різнобічну інформацію, і, першою чергою, ідентифікувати речовини, визначити їх кількість, встановити структуру біологічно активних сполук, а також вивчити механізми біохімічних реакцій. Фотоколориметрія– це вимірювання поглинання видимої частини спектру забарвленими розчинами. Для наукових досліджень фотоколориметрія застосовується при визначенні активності ферменту супероксиддисмутази, загальної антиоксидантної активності плазми крові та еритроцитів, гідропероксидів ліпідів плазми крові, серомукоїдів сироватки крові тощо. Власне спектрофотометрія – це вимірювання поглинання (і пропускання) прозорих розчинів в ультрафіолетовій, видимій та інфрачервоній зонах спектру (220 – 1 100 нм). Нефелометрія – це метод аналізу, пов’язаний із оцінкою ступеня каламутності досліджуваного розчину. Інтенсивність розсіювання залежить від розмірів частинок і кількості розчиненої речовини. Імунофлюоресценція – зручний метод аналізу багатьох зразків на вміст та ідентифікацію специфічних антитіл. Широкого застосування набув метод імунофлюоресценції в наукових дослідженнях для оцінки клітин і зразків тканин. Електрофорез – це метод розділення заряджених частинок у електричному полі. У даний час метод електрофорезу широко використовують для фракціонування білків і ліпопротеїнів. Хроматографічні методи. Для розділення та кількісного визначення білків і амінокислот, нуклеїнових кислот, вуглеводів, ліпідів та інших метаболітів використовують різні методи хроматографіїПолярографія – це електрохімічний метод, в основі якого лежить автоматична реєстрація сили струму при поступовому збільшенні напруги на електродах, занурених у досліджуваний розчин. Полярографія – це електрохімічний метод, в основі якого лежить автоматична реєстрація сили струму при поступовому збільшенні напруги на електродах, занурених у досліджуваний розчин. Під час проходження струму в процесі електровідновлення або електроокиснення на полярограмах з’являються ділянки, на яких сила струму пропорційна концентрації реагуючих речовин. Зміна висоти полярограм дає уявлення про концентрацію речовини. Імуноферментний аналіз (ІФА). У біохімії широко використовують методи, які поєднують імунні реакції з іншими фізико-хімічними методами – хроматографічні методи в імунології, імунофлюоресцентний аналіз, імуноелектрофорез і радіоімунний аналіз. Полімеразна ланцюгова реакція (ПЛР) застосовується у діагностиці захворювань, що передаються статевим шляхом. Реакція дозволяє множити певні нуклеотидні послідовності до кількостей, які можна виявити методом молекулярної гібридизації за допомогою електрофорезу. ПЛР є багатократно повторюваними циклами синтезу (ампліфікації) специфічної ділянки ДНК-полімерази, дезоксинуклеозидтрифосфатів, відповідного сольового буферу та олігонуклеотидних затравок - праймерів, які визначають межі ампліфікованої ділянки ДНК-мішені. ПЛР застосовується у клініці для діагностики гонореї, трихомоніазу, цитомегаловірусу, мікоплазмозу, уреаплазмозу, герпесу тощо
Мета проведення біохімічних лабораторних досліджень і критерії оцінки використаних методів лабораторних досліджень. До способів розрахунку результатів дослідження, які використовують у клінічній біохімії, можна віднести застосування умовних одиниць, розрахунки за стандартним (еталонним розчином), калібрувальним графіком, коефіцієнтом перерахунку. Умовні одиницівикористовуються поряд із системними одиницями переважно в традиційних методиках. Стандартні розчини обробляють одночасно із серією досліджуваного біоматеріалу і перебувають у тих самих умовах, що й останній. Еталонні розчини повинні містити точно визначену концентрацію певної речовини. Результати дослідження розраховують за пропорцією: за трьома відомими величинами встановлюється четверта, досліджувана: Аст Сст ______ = ______ , С х= Адос ∙ Сст / Аст Адос Сх де Аст – оптична густина стандартного розчинуАдос – оптична густина досліджуваної проби; Сст – відома концентрація стандартного розчину;Сх – досліджувана концентрація проби. Калібрувальні графіки будують для кожного фотометра, використовуючи серію стандартних розчинів. Такі розчини різняться за концентрацією речовини у порядку наростання. Розрахунок за допомогою коефіцієнта перерахунку найпростіший. Розрахунки виконують за формулою: k × Адос
Матеріал для лабораторних діагностичних досліджень, принципи забору та збереження матеріалу для лабораторних досліджень. Мокрота. Мокрота збирається в чисту суху широкогорлу склянку з кришкою, що закручується, при кашлі, а не при відхаркуванні, про що треба попередити хворого. Для запобігання попадання в мокроту домішок з порожнини рота перед виділенням мокроти хворий повинен старанно прополоскати рот водою. Зберігати зібрану мокроту найкраще в холодильнику або в прохолодному місці, як можна найкоротший час. Джерелом найбільш цінної інформації є вміст трахеобронхіального дерева, отриманий при бронхоскопії (промивні води бронхів). Ексудати та трансудати. Їх одержують шляхом пункції серозних порожнин, збирають у чисту пробірку, добавляють цитрат натрію (1 г на 1 л рідини) або гепарин ex tempore і всю кількість відправляють у лабораторію на дослідження. Спиномозкову рідину (ліквор) одержують при люмбальній пункції. Як правило, для аналізу ліквору достатньо8-10 мл. спино-мозкової рідини, яку зразу ж доставляють до лабораторії. Кістковий мозок. Спочатку проводиться пункція грудини або подвздовшньої кістки,робляться мазки пунктату на предметних скельцях, після чого проводяться цитологічні дослідження. Крім того, роблять трепабіопсію подвздовшньої кістки з наступним гістологічним дослідженням зрізів кісткового мозку. Пункцію проводять у процедурному кабінеті з дотриманням правил асептики. Щоб одержати якісні результати дослідження, необхідно пам'ятати, що інструменти повинні бути старанно знезводжені (вода ушкоджує клітинні елементи). З огляду на швидку здатність пунктату до згортання, останній після одержання необхідно зразу ж розвести фізіологічним розчином хлористого натрію (до підрахунку каріоцитів), після чого варто швидко приготувати мазки. Подальше опрацювання мазків пунктату проводять так само, як і мазків крові. Лімфатичні вузли. При морфологічному дослідженні лімфатичних вузлів виконується пункція, з подальшим проведенням цитологічного аналізу пунктату, а також біопсія з наступним гістологічним вивченням зрізів. Пункція є найбільше доступною процедурою, може проводитися водночас у вузлах різноманітної локалізації, а також повторно.Невелику кількість вмісту лімфатичного вузла видавлюють з шприца на предметне скло і готують тонкі мазки. Якщо в пунктаті виявиться значна кількість домішків крові, рекомендується вибрати більш світлі крупинки і розмазати їх тонким прошарком по предметному склі або готувати препарати за принципом відбитків. Найбільшу інформацію дає поєднане дослідження: біопсія лімфатичного вузла, приготування відбитків на предметному склі для цитологічного та направлення препарату для гістологічного досліджень. Мікробіологічні дослідження. Збір проб для мікробіологічних досліджень необхідно проводити: а) до початку антибактеріальної терапії; б) безпосередньо з осередку інфекції; в) на початку ознобу та на висоті температурної реакції; г) стерильними ватяними тампонами або шприцом внести матеріал в стерильний посуд з дотриманням найсуворішої асептики; (е) для виділенняанаеробів перед пункцією з шприца попередньо видалити повітря, а після пункції зразу ж закрити голку ковпачком; при дослідженні шматочків тканини їх беруть із глибини осередку патологічного утворення; при використовуванні тампонів для забору біологічного матеріалу, останнівідразу требазанурювати в транспортне середовище.
Характеристика помилок, що мають місце під час проведення лабораторних досліджень. У ході лабораторного дослідження можна припуститися помилок. До складу остаточного результату кожного визначення входять остаточно дійсна вартість (дійсні величини) та певні помилки. Оцінка достовірності результату та його клінічна оцінка потребують знання видів помилок. Загалом під час діагностичних досліджень трапляються помилки, які можна класифікувати так: 1) помилка перед проведенням дослідження; 2) аналітична лабораторна помилка; 3) інтерпретаційна помилка. Помилка перед проведенням дослідження охоплює групу факторів, які можуть впливати на остаточний результат досліджень, доки матеріал аналізують у лабораторії. Ці фактори пов’язані з підготовкою хворого до дослідження, із забором та зберіганням матеріалу до початку аналізу. Аналітична (лабораторна) помилка. Ця помилка пов’язана з ходом дослідження біологічного матеріалу в лабораторії. Систематична помилка (точності) відтворює різницю між величиною отриманою і дійсною. Випадкова помилка (повторюваності) забезпечується різницею між результатами вимірювання одного й того ж самого зразка на тому самому лабораторному устаткуванні й тим самим методом, спричиненою змінами умов під час виконання вимірів. Джерело випадкової помилки пов’язане зі взаємодією багатьох непередбачуваних факторів і тих випадків, які змінюють умови визначення. Саме ця зміна зумовлює різницю результатів кількох визначень одного й того ж самого зразка в тому самому матеріалі.
Ферменти: визначення; властивості ферментів як біологічних каталізаторів реакцій обміну речовин. Ферменти - високоспеціалізовані універсальні за своєю біологічною функцією білкові сполуки, які синтезуються в процесі життєдіяльності всіх живих організмів і виконують функції каталізаторів біохімічних процесів, тобто прискорюють і здійснюють перебіг певних хімічних реакцій у живому організмі. Ферменти можуть виявляти свою активність як в організмі, так і будучи виділеними з нього. Значення ферментів для життєдіяльності величезне. У присутності ферментів хімічні процеси в організмі перебігають дуже швидко (інколи за мільярдні долі секунди). Без ферментів ці ж хімічні реакції відбувалися б у мільйони разів довше, що не сумісно з тим рівнем обміну речовин і енергії, який властивий живій матерії. Наприклад, в організмі людини за одну секунду руйнується і синтезується 3 мільйони еритроцитів. До складу одного еритроцита входить 300 -400 мільйонів молекул гемоглобіну – гемпротеїну з молекулярною масою 65 000 – 68 000 Да. Звідси можна уявити собі швидкість хімічних реакцій у живому організмі, які забезпечують ці процеси. На даний час охарактеризовано декілька тисяч ферментів, понад тисячу з них отримані в хімічно чистому вигляді. Для багатьох білків-ферментів з'ясована їх амінокислотна послідовність. Вивчення ферментів має величезне значення для будь-якої фундаментальної і прикладної галузі біології, для багатьох практичних галузей хімічної, харчової і фармацевтичної індустрії, зайнятих приготуванням каталізаторів, антибіотиків, вітамінів, амінокислот, пептонів та інших біологічно активних речовин, які використовуються в народному господарстві та медицині. Крім цього, ферменти широко використовуються в науково-дослідних роботах як високоспецифічні реагенти, як інструменти при з'ясуванні будови біополімерів (наприклад, нуклеїнових кислот), при генно-інженерних розробках тощо. В останні роки вчення про ферменти відокремилося в окрему галузь біохімічної науки – ферментологію (ензимологію), яка розвивається в тісному зв'язку з багатьма науками, зокрема, з органічною, неорганічною та фізичною хіміями, фізіологією, токсикологією, мікробіологією, генетикою, фармакологією тощо. Таким чином, ця галузь знань знаходиться на стику хімічних, біологічних і медичних наук.
Фізико-хімічні властивості білків-ферментів: електрохімічні (поверхневий заряд молекули) властивості, розчинність, термодинамічна стабільність білкових молекул-ферментів, осадження, денатурація, взаємодія з лігандами та її функціональне значення. На сьогоднішній день достеменно відомо, що ферменти – це речовини білкової природи. Підтвердженням цього є факт втрати ферментами бродіння своєї активності під час кип’ятіння, що було досліджено ще Л.Пастером. Кип’ятіння спричинює незворотну денатурацію білка-фермента, внаслідок чого останній втрачає здатність каталізувати хімічну реакцію. Як відомо, білки теж при кип’ятінні денатурують і втрачають сої біологічні властивості. Дія на ферменти різних фізичних і хімічних чинників, таких як вплив УФ- і рентгенівського опромінення, ультразвуку, осадження мінеральними кислотами, лугами, алкалоїдними реактивами, солями тяжких металів тощо теж спричинює денатурацію ферментів, так само як і білків, і втрату їх каталітичної активності. В основі денатурації лежить руйнування зв’язків, що стабілізують вищі структури ферменту-білка (четвертинну, третинну, вторинну) і як наслідок випадання його в осад. Це свідчить про те, що просторова структура білка впливає на виявлення його ферментативної активності. Фізико – хімічні властивості перелічених амінокислотних залишків поліпептидного ланцюга ферменту визначають контакт із відповідним субстратом та його перетворення. Гідрофобні радикали амінокислот мають спорідненість до неполярних ділянок субстрату. Полярні групи проявляють кислотні, або основні, або спряжені кислотно-основні (наприклад, гістидин) властивості. Зсув рН середовища викликає зміни їх кислотно-основних властивостей і сприяє контакту з різними групами субстрату. Із препаративною метою часто обезводнюють ферменти (видаляють воду) у вакуумі з замороженого розчину (ліофілізація). Осадження з розчину ферментів спиртом чи ацетоном теж здійснюють при низькій температурі, оскільки при кімнатній температурі ці процедури спричинюють втрату ферментативної активності. Для стабілізації ферментів часто користуються хелатоутворювальними агентами, наприклад, ЕДТА (етилендіамінтетраацетат), який може зв’язувати небажані домішки, які гальмують активність фермента. Однією з обов’язкових умов збереження стабільності фермента є його зберігання в висушеному або замороженому стані, велика кількість ферментів може зберігати свою стабільність у вигляді суспензії в концентрованих розчинах амонію сульфату.
Прості та складні білки-ферменти. Кофактори, коферменти та простетичні групи складних ферментів. Навести приклади. Роль іонів металів у функціонуванні ферментів. ферменти – це білки, що проявляють каталітичні властивості, тобто вони прискорюють перебіг різних хімічних процесів, які відбуваються в живому організмі. Ферментам притаманні всі фізико-хімічні властивості білків: висока молекулярна маса, розщеплення до амінокислот під час гідролізу, утворення колоїдоподібних розчинів; вони не стійкі до впливу високих температур та солей важких металів, проявляють антигенні властивості, піддаються фракціонуванню Ферменти-прості білки являють собою поліпептидні ланцюги, які при гідролізі розпадаються до амінокислот, їх ще називають однокомпонентними. До них належать пепсин, трипсин, уреаза, рибонуклеаза тощо. Ферменти-складні білки крім поліпептидних ланцюгів містять небілкову частину, їх називають двокомпонентними. Білкову частину двокомпонентних ферментів називаютьапоферментом (забезпечує специфічність дії та відповідає за вибір типу хімічного перетворення субстрату), а небілкову –кофактором (слугує акцептором і донором хімічних груп, атомів і електронів у каталітичній ділянці активного центра фермента). Молекула складного фермента в цілому називається холоферментом.Зв'язок білкової частини ферменту з небілковою здійснюється за рахунок ковалентних і нековалентних зв’язків і може бути різної міцності. Якщо небілкова частина ферменту міцно пов'язана з білком і в циклі біохімічних реакцій не відділяється від нього, її прийнято називати простетичною групою(наприклад, ФАД, ФМН, біотин тощо). Небілкові компоненти, які слабо пов'язані з білком і легко дисоціюють з комплексу з ферментним білком, називають коферментами (наприклад, НАД+, НАДФ+). Коферменти можуть знаходитися у вільному стані й сполучатися з білковою частиною тільки в момент каталітичної реакції, їх можна розглядати в якості другого субстрату. Один і той самий кофермент може сполучатися з різними апоферментами і брати участь у різних хімічних перетвореннях субстрату (наприклад, піридоксальфосфат може брати участь у реакціях трансамінування чи декарбоксилування). Ферменти, які міцно пов’язані з іонами металів і не втрачають цього зв’язку за умов виділення та фракціонування ферменту, називаютьсяметалоферментами. У деяких випадках іони металів не входять до складу ферментів як інтегральні структурні компоненти, а виконують лише функцію їх активаторів (іони Са2+ служать активаторами протеїнкінази С, іони Сl- – a-амілази тощо).
Будова ферментів: активний, регуляторний (алостеричний) центри, їх значення. Більшість ферментів має чотири рівні структурної організації (первинну, вторинну, третинну і четвертинну), тобто є олігомерними білками, що складаються із протомерів. Кожна із субодиниць або окремі їх частини відіграють певну роль у процесі функціонування ферменту. Прості (однокомпонентні) ферменти здійснюють ферментативне перетворення субстрату з участю власне білкової молекули. Безпосередню участь у реакції бере не весь поліпептидний ланцюг ферменту, а тільки незначна його частина, що близько прилягає до субстрату. У ферментативну реакцію включається тільки декілька залишків амінокислот. Ці залишки можуть розташовуватися в поліпептидному ланцюзі як поруч, так і далеко один від одного, але просторово вони повинні бути досить зближені. Та частина молекули ферменту, яка з'єднується із субстратом, називається активним центром ферменту. Активний центр відповідає за специфічну спорідненість ферменту із субстратом, утворення ферменто-субстратного комплексу і каталітичне перетворення субстрату. В активному центрі ферменту умовно розрізняють так звану каталітичну ділянку, де відбувається каталітичне перетворення субстрату, і контактну, або якірну ділянку, що зв'язує фермент із субстратом. За утворення активного центру ферменту, як і за його каталітичну дію, відповідає третинна структура білкової молекули. Отже, при порушенні третинної структури (денатурація) роз'єднуються просторово поєднані амінокислотні залишки і, як наслідок, фермент втрачає активність. У складі активного центру простого ферменту знаходиться приблизно 15 залишків амінокислот. Активний центр утворюють залишки таких амінокислот, як серин, цистеїн, гістидин, тирозин, лізин та деякі інші, що надають ферменту як просторової, так і електричної спорідненості із субстратом. В утворенні тимчасового комплексу між ферментом і субстратом важлива роль належить дисульфідним, іонним, а також слабким зв'язкам (водневі зв'язки, гідрофобна взаємодія). В утворенні активних центрів беруть участь також кофактори даного ферменту: простетичні групи, іони металів. Активний центр складних (двокомпонентних) ферментів містить у своєму складі як кофермент, так і ту частину апоферменту, що просторово прилягає до нього. Кофермент при цьому може відповідати за утворення зв'язку із субстратом, формування третинної або четвертинної структури апоферменту і каталітичне перетворення субстрату. Ферменти можуть мати 1, 2, 3 і більше активних центрів, що залежить від кількості протомерів (субодиниць), які входять у його структуру. Крім активних центрів, у ферментах можуть бути ще так звані алостеричні центри (від грец. алос — інший, другий; стереос — просторовий, структурний). Алостеричні центри служать місцем впливу на фермент різних регуляторних чинників, тому їх ще називають регуляторними центрами, а речовини, що взаємодіють з алостеричним центром, отримали назву ефекторів. Приєднання до алостеричного центру ефектора призводить до певних структурних змін в активному центрі та, як наслідок, пригнічення або підвищення активності ферменту. Ефекторами можуть служити продукти ферментативних реакцій, гормони, медіатори нервової системи, метали. Алостеричних центрів (як і активних) фермент може мати декілька, відповідно до кількості протомерів. Важливо зазначити, що алостеричні й активні центри у ферментах просторово відокремлені, тобто знаходяться один від одного на певній відстані. Механізми дії ферментів Ферменти збільшують швидкості біохімічних реакцій, які вони каталізують, у 108-1020 разів; при відсутності ферменту будь-яка метаболічна реакція практично не відбувається. Відомо, що константа швидкості хімічної реакції залежить від її енергії активації та температури, що виражається рівнянням Арреніуса в експоненційній формі: k = Ае-∆Е/RT. Під енергією активації (∆Е в рівнянні Арреніуса) в хімічній термодинаміці розуміють додаткову енергію, необхідну для переходу молекул (субстратів S) у перехідний (активований) стан (S*), який передує їх перетворенню в продукти реакції. Згідно з цим, експоненційний член рівняння е-∆Е/RT (фактор Больцмана) - частка молекул у системі, які мають енергію, достатню для хімічного перетворення. Оскільки всі метаболічні процеси в живих організмах перебігають в ізотермічних умовах, каталітична дія ферментів реалізується за рахунок зниження енергії активації (∆Е) біохімічної реакції, що збільшує фактор Больцмана і, відповідно, константу швидкості реакції на декілька порядків.
Рівні структурної організації ферментів. Мультиферментні комплекси, ферментативні ансамблі, поліфункціональні ферменти, їх переваги. Більшість ферментів має чотири рівні структурної організації (первинну, вторинну, третинну і четвертинну), тобто є олігомерами, які складаються з протомерів. Кожна із субодиниць або окремі їх частини відіграють певну роль у процесі функціонування ферменту. Прості (однокомпонентні) ферменти здійснюють ферментативне перетворення субстрату з участю власне білкової молекули. Безпосередню участь у реакції бере не весь поліпептидний ланцюг ферменту, а тільки незначна його частина, що близько прилягає до субстрату – активний центр. У ферментативну реакцію включається тільки декілька просторово наближених до нього залишків амінокислот. За утворення активного центру ферменту, як і за його каталітичну дію, відповідає третинна структура білкової молекули. Отже, при порушенні третинної структури (денатурація) роз’єднуються просторово розміщені амінокислотні залишки і, як наслідок, фермент втрачає активність. Активний центр складних (двокомпонентних) ферментів, як відомо, містить у своєму складі як кофермент, так і ту частину апоферменту, що просторово прилягає до нього. Кофермент при цьому може відповідати за утворення зв’язку із субстратом, формування третинної або четвертинної структури апоферменту і каталітичне перетворення субстрату.Кожна клітина організму має свій специфічний набір ферментів. У клітині дія кожного ферменту не індивідуальна, а тісно пов’язана з іншими ферментами. Так із окремих ферментів формуються поліферментативні (мультиферментативні) комплекси, що каналізують послідовності спряжених біохімічних реакцій. Можна умовно виділити наступні види організації поліферментативних систем: функціональна, структурно-функціональна і змішана. Функціональна організація характеризується тим, що окремі ферменти, об’єднанні в поліферментну систему, виконують певну функцію, з допомогою метаболітів, які дифундують від одного ферменту до іншого. Причому в функціонально організованих поліферментативних системах продукт реакції першого ферменту в ланцюгу служить субстратом для наступного тощо. Часто метаболіт може виконувати об’єднуючу роль між різними поліферментними системами, якщо він є загальним для індивідуальних ферментів різних поліферментативних ланцюгів, кожен з яких виконує свої біохімічні функції в клітині. Такі метаболіти об’єднують ферментативні системи навіть локалізовані в різних органоїдах клітини в єдину метаболічну карту клітини. Прикладом функціональної організації поліферментної системи служить гліколіз, тобто розпад глюкози. Всі ферменти гліколізу знаходяться в розчиненому стані. Кожна реакція каталізується окремими ферментами. Зв’язуючою ланкою тут виступають метаболіти. Положення кожного ферменту в ланцюгу встановлюється за їх спорідненістю до субстрату.Структурно-функціональна організація полягає в тому, що ферменти утворюють структурні системи з певною функцією при допомомозі фермент-ферментативних (білок-білкових) взаємодій. Таким чином формуються поліферментативні надмолекулярні комплекси. До них відноситься, наприклад, поліферментний комплекс піруватдегідрогеназа, який складається із декількох ферментів, що беруть участь в окисненні пірувату, або синтетаза жирних кислот, яка складається із семи структурно зв’язаних ферментів. Такі поліферментативні комплекси досить міцні і важко розпадаються на окремі ферменти.Низка таких мультиферментативних комплексів іноді називають ферментними ансамблями, які структурно пов’язані з певними органелами (рибосоми, мітохондрії) або з біомембраною і становлять високоорганізовані надмолекулярні системи (наприклад, тканинне дихання, пов’язане з перенесенням електронів від субстрату до кисню через систему дихальних ферментів). Змішаний тип організації поліферментативних систем становить комбінацію обох типів організації, тобто частина поліферментної системи має структурну організацію, частина – функціональну (такі ферменти називають поліфункціональними). Прикладом такої організації служить поліферментна система циклу Кребса, де частина ферментів об’єднана в структурний комплекс (альфа-кетоглутаратдегідрогеназний комплекс), а частина з’єднана одна з одною функціонально за допомогою метаболітів.
Номенклатура та класифікація ферментів. Типи реакцій, що каталізують окремі класи ферментів. Шифр ферментів. Висока специфічність дії ферментів зумовлює дуже велику їх кількість. Майже кожна реакція, що відбувається в живому організмі, здійснюється за участю специфічно пристосованого до неї ферменту або групи ферментів. На даний час відомо біля 2000 різних ферментів, і кількість їх продовжує збільшуватися.Всі відомі реакції, що відбуваються в організмі, були поділені на шість основних типів. Відповідно до шести типів хімічних реакцій усі відомі ферменти діляться на шість класів:1. Оксидоредуктази - ферменти, що каталізують окисно-відновні реакції, тобто перенесення електронів і атомів водню від однієї речовини (донатора) до іншої (акцептора). У результаті реакцій, що каталізуються оксидоредуктазами, клітини одержують хімічну енергію.Оксидоредуктази - це складні ферменти. Кофакторами в їх складі можуть бути нікотинамідні коферменти (НАД і НАДФ), флавіннуклеотиди (ФМН і ФАД), залізопорфіринові комплекси (гем) і багато катіонів (Fe2+, Fe2+, Cu2+, Zn2+ і ін.). Зазначені кофактори спроможні виконувати роль акцепторів електронів і атомів водню і передавати їх іншим речовинам.2. Трансферази - клас ферментів, що каталізують транспорт різноманітних хімічних груп від однієї речовини (донатора) до іншого (акцептора).3. Гідролази - ферменти, що каталізують реакції розщеплення речовин за участю води. Каталітична активність цих ферментів залежить від наявності в їх каталітичному центрі HS-груп. Гідролази дуже поширені в природі і беруть участь в обміні вуглеводів, жирів, білків та інших сполук. 4. Ліази - ферменти, що каталізують реакції негідролітичного відщеплення певних груп з утворенням подвійних зв'язків або приєднання групи в місці подвійного зв'язку. Ці реакції здійснюються без використання енергії макроергичних сполук. Ліази, як правило - складні ферменти, що містять як кофактори фосфорні ефіри водорозчинних вітамінів.5. Ізомерази - ферменти, що каталізують реакції внутрішньомолекулярного переміщення різних груп або реакції утворення ізомерів. Типовим прикладом ферментів цього класу є тріозофосфатізомераза. Даний фермент зворотно перетворює 3-фосфогліцериновий альдегід у діоксіацетонфосфат. 6. Лігази (синтетази) - ферменти, за участю яких здійснюється приєднання одна до одної двох молекул із використанням енергії АТФ і утворенням нових зв'язків. Лігази називають ще синтетазами, оскільки вони є каталізаторами синтетичних реакцій. Це складні ферменти. Кофактором багатьох із них є вітамін Н, або біотин. Відповідно до нової номенклатури назви ферментів складаються з двох частин. Перша частина вказує на назву субстрату, друга - на природу хімічної реакції, яку здійснює фермент, із додаванням суфіксу -аза. Якщо фермент каталізує реакцію між двома субстратами, то в першій частині назви ферменту дають назви обох субстратів, розділених двома крапками, в другій — вказують тип хімічної реакції з додаванням суфіксу -аза.
Специфічні властивості ферментів: залежність активності ферментів від рН середовища, температури (пояснити і зобразити графічно). Однак, для ферментів характерні і специфічні властивості, які відрізняють їх від інших каталізаторів. Ці відмінності пов'язані з особливостями будови ферментів, які є складними білковими молекулами: 1. ефективність ферментів вища, ніж небілкових каталізаторів: швидкість перебігу реакції за участю ферментів зростає в 108 – 1020 разів (фермент уреаза прискорює гідроліз сечовини в 1014 разів), вони діють у мізерних концентраціях (молекула реніну, який синтезується в шлунку теляти, звурджує за 10 хв при температурі 37ºС 106 молекул казеїногену). 2. Ферменти мають високу специфічність дії, яка обумовлена унікальною структурою активного центра, а також конформаційною та електростатичною комплементарністю між молекулами субстрату та фермента. Кожний фермент каталітично прискорює, зазвичай, одну хімічну реакцію, або ж групу реакцій одного типу. Висока специфічність дозволяє ферментам брати участь у регуляції обміну речовин і направляти його в певному напрямі. 3. Активність ферментів може суттєво змінюватися під впливом сполук, котрі прискорюють (активатори) та сповільнюють (інгібітори) швидкість каталізованої реакції, що дає можливість координувати метаболічні процеси, спрямовані на відтворення живої матерії, підтримання постійності гомеостазу та пристосування до умов середовища. 4. Термолабільність ферментів: ферменти каталізують хімічні реакції при невисокій температурі (37 – 40°С); її зростання призводить до денатурації білкової молекули фермента та, відповідно, зниження швидкості реакції або повної її зупинки. При температурі 100°С майже всі ферменти втрачають свою активність. зниження температури нижче оптимальної тимчасово сповільнює активність фермента внаслідок зменшення швидкості дифузії молекул. 5. залежність активності ферментів від рн середовища: ферменти зазвичай найактивніші в межах вузької концентрації іонів Н+ (фізіологічне значення рН = 6,0 – 8,0). Виключення становлять пепсин (оптимум рН = 2,0) та аргіназа (оптимум рН = 10,0). Зміна рН середовища впливає на стан і ступінь іонізації кислотних і основних груп, які входять до складу фермента в цілому та його активного центра зокрема, що впливає на третинну структуру білка та формування активного фермент-субстратного комплексу. 6. У випадку, коли ферментативний процес являє собою низку послідовних реакцій (метаболічні шляхи) дія ферментів строго впорядкована: продукт однієї ферментативної реакції слугує субстратом для іншої. Їх активність змінюється в залежності від потреб організму в кінцевому продукті.
Специфічність ферментів. Види специфічності (абсолютна, відносна, стереоспецифічність). Навести приклади. Специфічність – одна з найважливіших властивостей ферментів, яка визначає біологічну значимість цих білкових молекул і істотно відрізняє їх від небілкових каталізаторів. В основі специфічності лежить відповідність стеричної структури субстрату й активного центра фермента, внаслідок чого даний фермент з безлічі речовин, які є в клітині, приєднує лише певний субстрат. Розрізняють субстратну та каталітичну специфічності фермента. Субстратна специфічність – це здатність фермента взаємодіяти з одним або кількома субстратами. Вона може бути абсолютною, груповою та стереоспецифічністю. Фермент з абсолютною субстратною специфічністю каталізує перетворення лише одного субстрату з певною структурою. Будь-які модифікації (зміни) у структурі субстрату роблять його недоступними для дії ферменту. Прикладом можуть слугувати аргіназа, яка каталізує реакцію розщеплення аргініну на сечовину та орнітин, та уреаза, яка каталізує гідроліз сечовини з утворенням вуглецю оксиду (ІІ) та аміаку. Сахараза гідролізує тільки сахарозу, а на інші дисахариди не діє. Переважна більшість ферментів каталізує однотипні реакції з невеликою кількістю (групою) структурно подібних субстратів із характерним типом зв’язку, тобто вони володіють груповою субстратною специфічністю. так, панкреатична ліпаза гідролізує жири в дванадцятипалій кишці, каталізуючи перетворення будь-якої молекули жиру до моноацилгліцеролу та двох молекул вищих жирних кислот, розриваючи ефірний зв'язок біля -атомів вуглецю гліцеролу, незалежно від того, які жирні кислоти входять до складу молекули жиру. Протеолітичні ферменти (пепсин, трипсин, хімотрипсин) теж володіють груповою субстратною специфічністю, гідролізуючи пептидні зв’язки, утворені різними амінокислотними залишками. Ферменти зі стереохімічною специфічністю діють лише на певні стереоізомери. Наприклад, більшість моносахаридів і продуктів їх обміну в організмі людини належать до D-стереоізомерів; ферменти, які здійснюють їх метаболізм, не володіють специфічністю до L-форм. Білки людини складаються з амінокислот L-ряду, тому більшість ферментів, які забезпечують перетворення амінокислот, володіють стереоспецифічністю до L-амінокислот. Якщо сполука існує в формі цис- і трансізомерів, то тільки одна з них може служити субстрат ом для дії ферменту, наприклад, фумараза каталізує перетворення фумарої кислоти (транс-ізомер), але не діє на малеїнову кислоту (цис-ізомер). Виключення становлять лише епімерази (рацемази), які каталізують перетворення оптичних ізомерів. Стереоспецифічність до - та -глікозидних зв’язків можна розглянути на прикладі амілази, яка діє лише на -глікозидні зв’язки, що дозволяє гідролізувати крохмаль і глікоген. Клітковина, хоч і теж складається з залишків глюкози, не гідролізується в організмі людини, оскільки містить у своєму складі -глікозидні зв’язки, для гідролізу яких в організмі людини немає відповідних ферментів. Каталітична специфічність – це такий вид специфічності, коли фермент каталізує перетворення приєднаного субстрату за одним із кількох можливих шляхів. Так, наприклад, молекула глюкозо-6-фосфату в гепатоцитах слугує субстратом для 4 різних ферментів: фосфоглюкомутази, глюкозо-6-фосфатфосфатази, фосфоглюкоізомерази та глюкозо-6-фосфатдегідрогенази. Завдяки особливостям будови каталітичних ділянок цих ферментів відбувається перетворення цього субстрату на 4 різних продукти
Внутрішньоклітинна локалізація ферментів. Навести приклади. Питання про локалізацію ферментів у клітині набуває особливої важливості, коли необхідно виділити фермент у чистому вигляді. Порівняно легко виявити локалізацію фермента методами цито- і гістохімії. Для цього тонкі зрізи тканин інкубують з відповідними субстратами, а після інкубації локалізацію продукту реакції досліджують за розвитком специфічної реакції при додаванні певних реактивів. У препаративній ферментології часто користуються методом диференційного центрифугування гомогенатів тканин. Для цього спочатку руйнують клітинну структуру з допомогою відповідного дезінтегратора і утворена гомогенізована маса підлягає диференційному центрифугуванню при температурі 0 - 4о С. Центрифугування різними швидкостями гомогенатів різних тканин забезпечує седиментацію (осідання) субклітинних структур, що дозволяє згодом дослідити їх склад (табл.1.2). Таблиця 1.2. Локалізація деяких ферментів усередині клітини. Наявність визначених ферментів у внутрішньоклітинних ділянках (компартментах) обумовлює функціонування в них метаболічних шляхів – перетворення одних метаболітів на інші.
Тканинна (органна) специфічність ферментів. Навести приклади. Відповідно, ферментний склад різних клітин різний. Ферменти, які виконують функцію життєзабезпечення, знаходяться в усіх клітинах організму. У процесі диференціювання клітин відбувається зміна їх ферментного складу. Так, фермент аргіназа який бере участь в утворенні сечовини, знаходиться лише в клітинах печінки, а кисла фосфатаза, яка бере участь у гідролізі моно ефірів ортофосфорної кислоти – у клітинах передміхурової залози. Це так звана органоспецифічність ферментів. Якщо говорити про вузькоспеціалізовані клітини, то ферментів, які забезпечують їх функціонування знаходиться в цих клітинах більше порівняно з іншими. Наприклад, у клітинах міокарда підвищена кількість креатинкінази та аспартатамінотрансферази, у гепатоцитах – аланінамінотрансферази, в остеобластах – лужної фосфатази тощо.
Класифікація коферментів за хімічною природою та за типом реакції, яку вони каталізують. Навести приклади. Як було сказано вище, для прояву каталітичної активності більшості ферментів необхідна присутність кофермента. Виключення становлять гідролітичні ферменти (протеази, ліпази, рибонуклеаза). Вони беруть участь в акті каталізу, здійснюють контакт між ферментним білком і субстратом, стабілізують апофермент, який, в свою чергу, посилює каталітичний акт небілкової частини і, крім цього, визначає специфічність ферментів, оскільки одна і та ж за хімізмом небілкова частина може функціонувати в складі різних ферментів. Хімічна природа коферментів, їх функції в ферментативних реакціях дуже різноманітні. Традиційно до коферментів відносять похідні вітамінів. Проте, окрім них є ще певна кількість небілкових сполук, які беруть участь у прояві каталітичної функції ферментів. За хімічною природою коферменти поділяють на:– похідні вітамінів (табл.1.1); – динуклеотиди (похідні нікотинаміду – НАД+, НАДФ+; похідне рибофлавіну – ФАД); – нуклеотиди – похідні пуринів та піримідинів (АТФ, АДФ, ЦТФ, ЦДФ, УТФ, УДФ); - комплекси порфіринів з іонами металів; - S-аденозилметіонін (SAM);– пептидні (глутатіон). Таблиця 1.1. Коферменти та відповідні їм вітаміни
Коферменти – переносники атомів водню та електронів (НАД+, НАДФ+ - коферменти – похідні вітаміну РР; ФАД, ФМН – коферменти – похідні вітаміну В2 – рибофлавіну; роль вітаміну С в окисно-відновних реакціях). (НАД+, НАДФ+, ФМН, ФАД, аскорбінова кислота, кофермент Q, глутатіон, гемінові коферменти (металопорфірини - цитохроми)). Похідні вітаміну РР – нікотинамідаденіндинуклеотид (НАД+) та нікотинамідаденіндинуклеотидфосфат (НАДФ+) це окиснені форми коферментів, у яких позитивний заряд несе атом азоту піридинового кільця нікотинаміду: Як видно зі схеми, субстрат (А) втрачає два атоми водню (2 протони та 2 електрони), але на кофермент переноситься лише 2 електрони і 1 протон, другий протон переходить у середовище. У результаті відновлена форма коферменту втрачає позитивний заряд. Нікотинаміддинуклеотиди входять до складу багатьох дегідрогеназ, необхідних для синтезу енергії в клітині: вони виступають акцепторами та проміжними переносниками атомів водню на початкових стадіях окиснення вуглеводів, жирних кислот, амінокислот, гліцерину, на стадії циклу Кребса і в термінальних стадіях дегідрування в дихальному та монооксигеназному ланцюгах; вони виступають у ролі алостеричних ефекторів ферментів енергетичного обміну. НАДФН2 як донор атомів водню використовується в біосинтетичних відновних реакціях (синтез жирних кислот, холестерину, гормонів тощо). Похідні вітаміну В2 – флавінмононуклеотид (ФМН) і флавінаденіндинуклеотид (ФАД) – коферменти, які входять до складу флавінових ферментів, що беруть участь у багатьох окиснювальних реакціях: перенесенні електронів і протонів у дихальному ланцюзі, окисненні пірувату, -кетоглутарату, жирних кислот, біогенних амінів тощо. Активною частиною флавінових коферментів є ізоалоксазинова циклічна система, вона може приєднувати два атоми водню (2 електрони та 2 протони): Флавінові коферменти, на відміну від нікотинамідних, зв’язують два протони. Вітамін С теж бере участь у окисно-відновних процесах. Він може існувати в двох формах – відновленій (аскорбінова кислота) та окисненій (дегідроаскорбінова кислота). Обидві форми легко переходять одна в одну і в якості коферментів гідроксилаз беруть участь в окисно-відновних реакціях: Ця властивість обумовлює участь аскорбінової кислоти в обміні білків, вуглеводів, мінеральних речовин. Металопорфірини за своєю структурою подібні або навіть тотожні гему в гемоглобіні. Порфіринові коферменти входять до таких ферментів як цитохроми (a, b, c), пероксидаза, каталаза тощо. Вони містять іони металів (зокрема заліза, міді тощо), які здатні змінювати свою валентність (наприклад, Fe2+ Fe3+) і, у такий спосіб, беруть участь у перенесенні електронів під час окисно-відновних процесів.


Коферменти – переносники хімічних груп (піридоксалеві коферменти; НS-КоА – коензим ацилювання; ліпоєва кислота; фолієва кислота). нуклеотидні - АТФ, АДФ УТФ, УДФ, ЦТФ, ЦДФ, похідні вітамінів - піридоксальфосфат, піридоксамінфосфат, ліпоєва кислота, КоА, тетрагідрофолієва кислота).Реакції за участю нуклеотидних коферментів зводяться до перетворення субстрату в молекулі коферменту (наприклад, перетворення УДФ-глюкози на УДФ-галактозу), вони можуть виступати донорами субстратів у реакціях перенесення тих чи інших груп (наприклад, УДФ-глюкоза є донором глюкози в процесі біосинтезу глікогену, ЦДФ-холін – донором холіну в біосинтезі холін фосфатидів тощо). Піридоксальфосфат і піридоксамінфосфат входять до складу багатьох ферментів у якості коферментів. Вони відіграють ключову роль в обміні амінокислот: каталізують реакції трансамінування (перенесення аміногрупи з -амінокислоти на -кетокислоту) та декарбоксилування (відщеплення карбоксильної групи у вигляді СО2) амінокислот, беруть участь у специфічних реакціях метаболізму окремих амінокислот (серину, треоніну, триптофану, сірковмісних амінокислот, а також у синтезі гему. Кофермент А (КоА) утворюється з пантотенової кислоти (В3), його сульфгідрильна група може зазнавати ацилування з перетворенням на ацил-КоА, або знаходитися в деацильованому стані (HS-КоА). Вони відіграють ключову роль в обміні амінокислот: каталізують реакції трансамінування (перенесення аміногрупи з -амінокислоти на -кетокислоту) та декарбоксилування (відщеплення карбоксильної групи у вигляді СО2) амінокислот, беруть участь у специфічних реакціях метаболізму окремих амінокислот (серину, треоніну, триптофану, сірковмісних амінокислот, а також у синтезі гему. Кофермент А (КоА) утворюється з пантотенової кислоти (В3), його сульфгідрильна група може зазнавати ацилування з перетворенням на ацил-КоА, або знаходитися в деацильованому стані (HS-КоА). Ці коферментні форми беруть участь у перенесенні ацильних радикалів у реакціях загального шляху катаболізму, активуванні жирних кислот, біосинтезі жирних кислот, холестерину, стероїдних гормонів, кетонових тіл, ацетилхоліну, знешкодженні чужорідних тіл у печінці. Тетрагідрофолієва кислота – відновлена форма ліпоєвої кислоти переносить одновуглецеві залишки (метильні (-СН3), оксиметильні (-СН2ОН), формільні (-НСО) тощо) на різні сполуки і бере тим самим участь у синтезі азотистих основ нуклеїнових кислот, креатину, метіоніну, гліцину, серину. Ліпоєва кислота завдяки своїй здатності легко переходити з окисненої у відновлену форми проявляє властивості коферменту в складі оксидоредуктаз, її амід слугує простетичною групою складного піруват- і -кетоглутаратдегідрогеназного комплексу, які беруть участь у окисненні піровиноградної та -кетоглутарової кислот.

Коферменти ізомеризації, синтезу та розщеплення С–С зв’язків (тіаміндифосфат; карбоксибіотин – біологічно активна форма вітаміну Н – біотину; метилкобаламін та дезоксиаденозилкобаламін – похідні вітаміну В12). (метилкобаламін, дезоксіаденозилкобаламін, карбоксибіотин, тіаміндифосфат). Біотин виконує коферментну функцію у якості N5-карбоксибіотину і входить до складу карбоксилаз: він бере участь в утворенні активної форми СО2, використовується в утворенні малоніл-КоА з ацетил-КоА, у синтезі пуринового кільця, у реакції карбоксилування пірувату з утворенням оксалоацетату тощо. Роль вітаміну В1 визначається тим, що у формі кофактора тіаміндифосфату (ТДФ) він входить до складу як мінімум трьох ферментів і ферментативних комплексів: у складі піруват- і -кетоглутаратдегідрогеназного комплексів він бере участь в оксиснювальному декарбоксилуванні пірувату та -кетоглутарату; у складі транскетолази він залучається у пентозофосфатний шлях перетворення вуглеводів. Тому цей кофермент необхідний для синтезу жирних кислот, холестерину, стероїдних гормонів, знешкодження токсичних речовин і ліків. Тіамінтрифосфат (ТТФ) у тканині мозку дотичний до синаптичної передачі нервових імпульсів. Метилкобаламін і дезоксіаденозилкобаламін – це коферментативні форми вітаміну В12. Метилкобаламін тісно пов’язаний із фолієвою кислотою, оскільки входить до складу фермента, який переносить метильну групу 5-метилтетрагідрофолієвої кислоти на гомоцистеїн із утворенням метіоніну. Разом із фолієвою кислотою він бере участь у синтезі креатину, азотистих основ, амінокислот, білків, нуклеїнових кислот тощо. Дезоксіаденозилкобаламін бере участь у завершальній стадії окиснення жирних кислот із непарною кількістю вуглецевих атомів, бічного ланцюга холестерину, тиміну, розгалужених амінокислот тощо.


Ізоферменти, визначення, особливості будови та функціонування.Значення в діагностиці захворювань ізоферментів лактатдегідрогенази і креатинфосфокінази. Ізоферменти (ізозоми) – це множинні генетично детерміновані форми одного ферменту, які каталізують одну й ту саму хімічну реакцію, але відрізняються за будовою, фізико-хімічними, імунологічними та каталітичними властивостями. Лише ферменти з четвертинною структурою можуть мати ізоферментні форми, які формуються в результаті комбінацій різних субодиниць. лактатдегідрогенази (ЛДГ )Цей фермент каталізує реакцію зворотного перетворення лактату в піруват: Для визначення активності ізоферментів ЛДГ використовують метод електрофорезуДля кожної тканини притаманне своє співвідношення ізоформ. Так, ЛДГ1 та ЛДГ2 переважають у органах, які характеризуються аеробним метаболізмом - серці, мозку, нирках, підшлунковій залозі та еритроцитах; ЛДГ3 – у легенях, селезінці, лімфатичних вузлах; ЛДГ4 та ЛДГ5 – в органах з активним анаеробним обміном – печінці та скелетних м’язах. Крім ізоферментів лактатдегідрогенази велике клінічне значення мають ізоферменти креатинфосфокінази (КК). Цей фермент є димером і побудований з двох основних субодиниць – В та М, з них утворюються три комбінації цих субодиниць і, як наслідок, три ізоферменти КК. У мозку переважає ізоформа ВВ, у скелетних м’язах – ММ, а у серцевому м’язі – ВМ. Визначення активності ізоферменту МВ найчастіше використовують для підтвердження діагнозу інфаркту міокарда. Підвищену активність цієї ізоформи виявляють вже на 4 – 6 годину після відчуття болю, пов’язаного з інфарктом, тоді як загальна активність КК може значно зростати при будь-якому пошкодженні скелетних м’язів (метаболічні порушення, запалення, ішемія тощо). Зростання активності ВВ-ізоформи супроводжує пошкодження мозку (крововилив у мозок, церебральний емболізм), деякі ракові захворювання. Проте, воно не має великого діагностичного значення.
Основні положення кінетики ферментативних реакцій: залежність швидкості реакції від концентрації субстрату, ферменту, рН та температури. Представити графічну залежність. Смислове значення величини Кm (спорідненість ферменту до субстрату). Ферментативна кінетика досліджує вплив на швидкість перебігу реакції різних хімічних речовин і фізико-хімічних чинників, які є достатньо численними та різноманітними. До них відносять концентрацію фермента та субстрату, рН і температуру, наявність активаторів або інгібіторів. Вивчення кінетики ферментативної реакції важливо для вибору одиниць активності ферментів, здійснення їх очищення, планування проведення досліджень та інтерпретації результатів тощо. Вплив концентрації фермента та субстрату на швидкість ферментативної реакції. Швидкість будь-якої ферментативної реакції залежить від концентрації фермента. У переважній більшості випадків у початковий період реакції, за умови надлишку фермента та невеликої кількості продукту швидкість реакції (V) прямо пропорційна його концентрації – [Е] і має лінійний характер: V=К×[Е], де К – коефіцієнт (рис. 5.4, а). Але з часом кількість продукту зростає і з’являється можливість для перебігу зворотної реакції, внаслідок чого лінійна залежність втрачається. Якщо ж концентрацію фермента залишити постійною, змінюючи лише концентрацію субстрату [S], то графік швидкості ферментативної реакції буде описуватися гіперболою. . Залежність швидкості ферментативної реакції від концентрації фермента (А) та субстрату (Б) При збільшенні кількості субстрату початкова швидкість зростає і коли фермент повністю насичується субстратом, тобто відбувається максимально можливе утворення фермент-субстратних комплексів, спостерігають найвищу швидкість утворення продукту. Але подальше збільшення концентрації субстрату не призведе до збільшення утворення продукту, оскільки швидкість реакції збільшуватися не буде. Описаний стан відповідає максимальній швидкості реакції (V max).

Графічний
і математичний вираз залежності
швидкості ферментативної реакції від
концентрації субстрату за Міхаелісом-Ментен
і Лайнуівером-Берком.
На основі аналізу залежності V від [S]
Л. Міхаеліс і М. Ментен сформулювали в
1913 р. загальну теорію кінетики дії
ферментів. Вони постулювали, зокрема,
що ферментативна реакція є двостадійною.
На першій стадії фермент вступає в
швидку зворотну взаємодію з субстратом
з утворенням фермент-субстратного
комплексу (ES), а під час другої стадії,
яка відбувається повільніше і лімітує
швидкість процесу, комплекс ES розпадається
з утворенням продукту реакції (Р) та
відновленого стану ферменту:
 де
к1
–
константа
швидкості утворення ES, к-1
– константа швидкості зворотної реакції
(розпаду ES), к2
–
константа
швидкості утворення продукту реакції.
Співвідношення констант швидкостей
(к-1
+ к2)
/ к1
називають
константою Міхаеліса (КМ).
На основі цього було виведено рівняння,
яке пов'язує V і [S], відоме під назвою
рівняння Міхаеліса:
де
к1
–
константа
швидкості утворення ES, к-1
– константа швидкості зворотної реакції
(розпаду ES), к2
–
константа
швидкості утворення продукту реакції.
Співвідношення констант швидкостей
(к-1
+ к2)
/ к1
називають
константою Міхаеліса (КМ).
На основі цього було виведено рівняння,
яке пов'язує V і [S], відоме під назвою
рівняння Міхаеліса: де:
V – початкова швидкість реакції, тобто
швидкість, що реєструється впродовж
періоду часу, за який рівень субстрату
не перевищує 10 %. У цей період швидкість
реакції можна вважати приблизно
постійною, оскільки, по-перше, зменшення
кількості субстрату невелике, а по-друге,
концентрація продукту незначна. Vмакс
дає характеристику каталітичній
активності фермента і має розмірність
швидкості ферментативної реакції
моль/л, тобто, вона визначає максимальну
можливість утворення продукту при
певній концентрації фермента в умовах
надлишку субстрату. У випадку, коли
швидкість реакції рівна половині
максимальної (V
= Vmax/2),
то КМ
=
[S]. Таким чином, константа Міхаеліса
чисельно дорівнює концентрації
субстрату, при якій швидкість
ферментативної реакції становить
половину від максимальної. Ця величина
характеризує спорідненість того чи
іншого фермента до конкретного субстрата
і є величиною постійною, незалежною
від концентрації фермента. Якщо КМ
значно більша від [S], тобто Км
>> S, то сума (КМ
+ S) приблизно дорівнює КМ,
відповідно рівняння набуває вигляду:
V = Vмакс
×
[S] / КМ.
у
цьому випадку швидкість ферментативної
реакції прямо пропорційна концентрації
субстрату, тобто при малих концентраціях
субстрату швидкість буде зростати і з
збільшенням концентрації. Якщо [S] >>
КМ,
то зростання концентрації субстрату
на величину КМ
+ [S] практично не впливає і нею можна
знехтувати. Тому швидкість реакції
буде дорівнювати максимальній швидкості:
V
= Vмакс.
Обробка рівняння Міхаеліса-Ментен за
методом подвійних зворотних величин
дає змогу відобразити залежність V від
[S] прямою лінією (рівняння Лайнуївера-Берка)
(рис. 5.5).
де:
V – початкова швидкість реакції, тобто
швидкість, що реєструється впродовж
періоду часу, за який рівень субстрату
не перевищує 10 %. У цей період швидкість
реакції можна вважати приблизно
постійною, оскільки, по-перше, зменшення
кількості субстрату невелике, а по-друге,
концентрація продукту незначна. Vмакс
дає характеристику каталітичній
активності фермента і має розмірність
швидкості ферментативної реакції
моль/л, тобто, вона визначає максимальну
можливість утворення продукту при
певній концентрації фермента в умовах
надлишку субстрату. У випадку, коли
швидкість реакції рівна половині
максимальної (V
= Vmax/2),
то КМ
=
[S]. Таким чином, константа Міхаеліса
чисельно дорівнює концентрації
субстрату, при якій швидкість
ферментативної реакції становить
половину від максимальної. Ця величина
характеризує спорідненість того чи
іншого фермента до конкретного субстрата
і є величиною постійною, незалежною
від концентрації фермента. Якщо КМ
значно більша від [S], тобто Км
>> S, то сума (КМ
+ S) приблизно дорівнює КМ,
відповідно рівняння набуває вигляду:
V = Vмакс
×
[S] / КМ.
у
цьому випадку швидкість ферментативної
реакції прямо пропорційна концентрації
субстрату, тобто при малих концентраціях
субстрату швидкість буде зростати і з
збільшенням концентрації. Якщо [S] >>
КМ,
то зростання концентрації субстрату
на величину КМ
+ [S] практично не впливає і нею можна
знехтувати. Тому швидкість реакції
буде дорівнювати максимальній швидкості:
V
= Vмакс.
Обробка рівняння Міхаеліса-Ментен за
методом подвійних зворотних величин
дає змогу відобразити залежність V від
[S] прямою лінією (рівняння Лайнуївера-Берка)
(рис. 5.5).
 Між
1/V
та
1/[S] є прямопропорційна залежність, яка
дозволяє легко отримати значення
кінетичних констант КМ
та
Vмакс,
що неможливо при аналізі звичайної
гіперболи. Із рівняння та графіка
випливає, що кутовий коефіцієнт прямої
(tg кута нахилу) дорівнює Км/Vмакс.
Значення цих констант легко знайти за
графіком. Залежність
швидкості ферментативної реакції від
температури. Зростання
температури до певних визначених меж
чинить вплив на швидкість ферментативної
реакції, подібно до впливу температури
на будь-яку хімічну реакцію, що
супроводжується прискоренням руху
молекул і, відповідно, прискоренням
ймовірності взаємодії реагуючих
речовин. Крім того, температура може
підвищувати енергію реагуючих речовин,
що теж прискорює реакцію. Однак, швидкість
ферментативної реакції має свій
температурний оптимум, перевищення
якого супроводжується зниженням
ферментативної активності внаслідок
термічної денатурації білкових молекул
(рис. 1.8). Для більшості ферментів людини
оптимальна температура 37 – 40 °С, проте
в природі існують і термостабільні
ферменти: Тaq-полімераза, виділена з
мікроорганізмів, яка не інактивується
навіть при 95 °С. Цей фермент використовують
у науково-практичній медицині для
молекулярної діагностики захворювань
із використанням методу ланцюгової
полімеразної реакції. Зниження
температури нижче оптимальної тимчасово
сповільнює активність ферменту внаслідок
зменшення процесів дифузії молекул;
повернення того чи іншого фермента в
оптимальне температурне середовище
відновлює його активність. Цю здатність
ферментів широко використовують у
медицині для пригнічення метаболічних
процесів у тканинах (під час трансплантації
органів, операцій на серці), у фармації
для збереження препаратів і лікарських
форм (наприклад, білкових препаратів,
відварів, настоїв, емульсій тощо), у
народному господарстві, наприклад, для
збереження харчових продуктів.
Залежність швидкості ферментативної
реакції від pH середовища.
Для
кожного фермента існує певне значення
рН середовища, в якому він виявляє
максимальну активність. Для пепсину
воно становить 1,5 – 2,0, піруваткарбоксилази
4,8, аргінази – 9,5 – 10,0. Проте, більшість
ферментів організму людини мають
оптимум рН, наближений до нейтрального:
фумараза 6,5; каталаза 6,8 – 7; уреаза 6,8 –
7,2; амілаза слини 6,8 – 7,4, карбоксипептидаза
7,5; трипсин 7,5 – 8,5, (рис. 1.9). Вплив рН на
активність фермента пов'язаний із
іонізацією функціональних груп
амінокислотних залишків білкової
молекули, що забезпечує оптимальну
конформацію активного центра. Відхилення
рН від оптимальних величин порушує
іонізацію функціональних груп в
активному центрі фермента. Так, наприклад,
залуження середовища спричинює
від’єднання іонів Н+
від карбоксильних груп (СОО-),
тоді як закислення, навпаки, приєднання
протонів до вільних аміногруп (NН3+).
Це викликає зниження активності
ферменту, порушує спорідненість
субстрату до фермента і гальмує
каталітичний процес в цілому. При
значному відхиленні від оптимального
значення рН може відбуватися денатурація
білкової молекули з повною втратою
ферментативної активності. Зміна рН
середовища може також впливати і на
просторову організацію субстрату.
Між
1/V
та
1/[S] є прямопропорційна залежність, яка
дозволяє легко отримати значення
кінетичних констант КМ
та
Vмакс,
що неможливо при аналізі звичайної
гіперболи. Із рівняння та графіка
випливає, що кутовий коефіцієнт прямої
(tg кута нахилу) дорівнює Км/Vмакс.
Значення цих констант легко знайти за
графіком. Залежність
швидкості ферментативної реакції від
температури. Зростання
температури до певних визначених меж
чинить вплив на швидкість ферментативної
реакції, подібно до впливу температури
на будь-яку хімічну реакцію, що
супроводжується прискоренням руху
молекул і, відповідно, прискоренням
ймовірності взаємодії реагуючих
речовин. Крім того, температура може
підвищувати енергію реагуючих речовин,
що теж прискорює реакцію. Однак, швидкість
ферментативної реакції має свій
температурний оптимум, перевищення
якого супроводжується зниженням
ферментативної активності внаслідок
термічної денатурації білкових молекул
(рис. 1.8). Для більшості ферментів людини
оптимальна температура 37 – 40 °С, проте
в природі існують і термостабільні
ферменти: Тaq-полімераза, виділена з
мікроорганізмів, яка не інактивується
навіть при 95 °С. Цей фермент використовують
у науково-практичній медицині для
молекулярної діагностики захворювань
із використанням методу ланцюгової
полімеразної реакції. Зниження
температури нижче оптимальної тимчасово
сповільнює активність ферменту внаслідок
зменшення процесів дифузії молекул;
повернення того чи іншого фермента в
оптимальне температурне середовище
відновлює його активність. Цю здатність
ферментів широко використовують у
медицині для пригнічення метаболічних
процесів у тканинах (під час трансплантації
органів, операцій на серці), у фармації
для збереження препаратів і лікарських
форм (наприклад, білкових препаратів,
відварів, настоїв, емульсій тощо), у
народному господарстві, наприклад, для
збереження харчових продуктів.
Залежність швидкості ферментативної
реакції від pH середовища.
Для
кожного фермента існує певне значення
рН середовища, в якому він виявляє
максимальну активність. Для пепсину
воно становить 1,5 – 2,0, піруваткарбоксилази
4,8, аргінази – 9,5 – 10,0. Проте, більшість
ферментів організму людини мають
оптимум рН, наближений до нейтрального:
фумараза 6,5; каталаза 6,8 – 7; уреаза 6,8 –
7,2; амілаза слини 6,8 – 7,4, карбоксипептидаза
7,5; трипсин 7,5 – 8,5, (рис. 1.9). Вплив рН на
активність фермента пов'язаний із
іонізацією функціональних груп
амінокислотних залишків білкової
молекули, що забезпечує оптимальну
конформацію активного центра. Відхилення
рН від оптимальних величин порушує
іонізацію функціональних груп в
активному центрі фермента. Так, наприклад,
залуження середовища спричинює
від’єднання іонів Н+
від карбоксильних груп (СОО-),
тоді як закислення, навпаки, приєднання
протонів до вільних аміногруп (NН3+).
Це викликає зниження активності
ферменту, порушує спорідненість
субстрату до фермента і гальмує
каталітичний процес в цілому. При
значному відхиленні від оптимального
значення рН може відбуватися денатурація
білкової молекули з повною втратою
ферментативної активності. Зміна рН
середовища може також впливати і на
просторову організацію субстрату.


Принципи кількісного визначення активності ферментів: за кількістю продукту, що утворюється під дією ферменту; за кількістю субстрату, що використовується; за зміною кількості коферменту (окисно-відновні перетворення для НАД та ФАД). Одиниці ферментативної активності. Здебільшого кількість фермента не можна виміряти в одиницях маси, тобто в міліграмах або в молях, про неї опосередковано свідчить його активність, тобто дія фермента. Іншими словами, присутність і кількість фермента виявляють за специфічністю та швидкістю реакції, яку він каталізує. Активність фермента можна визначити опосередковано: за визначенням кількості продукту, який утворився під дією ферменту, або за кількістю витраченого субстрату за одиницю часу при оптимальних умовах ферментативної реакції. Як приклад розглянемо принцип методу визначення активності -амілази, яка каталізує гідролітичне розщеплення крохмалю з утворенням у кінцевому результаті мальтози. Активність амілази розраховують на основі кількості негідролізованого крохмалю або за кількістю утвореної мальтози.Найчастіше активність ферментів визначають у сироватці, плазмі або клітинах крові та в сечі колориметричними, спектрофотометричними, флюориметричними, манометричними, кондуктометричними, віскозиметричними методами. У клініко-діагностичних лабораторіях для визначення активності ферментів переважно застосовують колориметричні та спектрофотометричні методи.В основі колориметричних методів лежить вимірювання за допомогою фотоелектроколориметра інтенсивності забарвлення речовини, яка утворюється під час взаємодії субстрату або продукту ферментативної реакції зі специфічними реактивами, які додаються у пробу після припинення реакції. Наприклад, продуктом дії ЛДГ в умовах прямої реакції є піруват, який з 2,4-динітро-фенілгідразином утворює у лужному середовищі забарвлений гідразон. Інтенсивність забарвлення пропорційна кількості пірувату, а значить, і активності фермента.Спектрофотометричні методи реєструють поглинання світла в певних ділянках спектра коферментами, субстратами або продуктами реакції. Ці методи широко застосовують для визначення активності оксидоредуктаз, зокрема НАД- та НАДФ-залежних дегідрогеназ. Перехід НАД+ або НАДФ+ з окисненої форми у відновлену супроводжується зміною поглинання в ультрафіолетовій ділянці спектра. В окисненій формі кофермент дає одну вузьку смугу поглинання при λ=260 нм, яка обумовлена наявністю аденіну в його молекулі. У разі відновлення кофермента поглинання світла в цій зоні дещо знижується, але з’являється інша широка смуга поглинання при λ=340 нм. Поява цієї смуги зумовлена зникненням одного подвійного зв’язку у піридиновому кільці аденіну під час його відновлення. Визначення активності ферментів за різницею спектрів поглинання окисненої та відновленої форм НАД+ (або НАДФ+) та НАДН (або НАДФН) називають тестом Варбурга (рис. 1.3)

Механізми дії ферментів (перетворення субстрату) утворення фермент-субстратного комплексу та процес перетворення субстрату. Ефекти зближення та орієнтації; кислотно-основного каталізу; нуклеофільного та електрофільного каталізу. Після того, як була встановлена хімічна природа ферментів, підтвердилися припущення англійського хіміка А. Бруна, висловлене ще на початку XX ст., а згодом і вчених В. Анрі, Л. Міхаеліса та М. Ментен про те, що в основі ферментативного каталізу лежить утворення нестійкого проміжного фермент-субстратного комплексу, який згодом розпадається з утворенням продуктів реакції та вивільненням фермента в незміненому вигляді. Ці твердження лягли в основу теорії «жорсткої матриці» Е.Фішера про те, що активний центр фермента комплементарний субстрату, тобто підходить до нього як «ключ до замка». Перша стадія – зближення та орієнтація субстрату відносно активного центра фермента з метою утворення фермент-субстратного комплексу – відбувається внаслідок зв’язування субстрату з активним центром ферменту водневими, електростатичними та гідрофобними взаємодіями, а в низці випадків – ковалентними та координаційними зв’язками. Друга стадія – перетворення первинного фермент-субстратного комплексу на один або кілька проміжних – характеризується послабленням зв'язків у субстраті, їх розривом або утворенням нових у результаті дії каталітичних груп фермента; формується молекула продукта. За рахунок утворення перехідних комплексів знижується енергія активації субстрату, що зумовлює зростання швидкості його перетворення. Ця стадія відбувається найповільніше та лімітує швидкість усього каталізу. Третя стадія – відокремлення від комплексу продуктів реакції, які утворилися в процесі ферментативної реакції та перехід фермента у початковий стан. Тривалість цієї стадії наближається до першої та визначається швидкістю дифузії продукту в середовище. Як уже зазначалося, ферменти прискорюють біохімічні реакції за рахунок зниження енергії активації. Енергія активації – це енергія, необхідна для переходу молекул 1 моля речовини в активний (перехідний) стан при певній температурі. Іншими словами, це енергія, необхідна для запуску хімічної реакції. Фермент знижує енергію активації шляхом збільшення числа активованих молекул, які стають більш реакційно здатними на нижчому енергетичному рівні. Прикладом такого каталізу може бути фермент алкогольдегідрогеназа печінки, яка містить іон n2+ та НАД+ у якості кофермента і каталізує реакцію окиснення етилового спирту: позитивно заряджений атом цинку сприяє від’єднанню протона від спиртової групи етанолу з утворенням негативно зарядженого атома кисню; негативний заряд перерозподіляється між атомом кисню та сусіднім атомом гідрогену, який потім у вигляді гідрит-іона переноситься на четвертий вуглецевий атом нікотинаміду з утворенням відновленої форми НАДН і оцтового альдегіду.

Регуляція ферментативних процесів шляхом зміни кількості ферментів (конститутивні та адаптивні ферменти) Регуляція швидкості ферментативної реакції здійснюється трьомах шляхами: зміною кількості ферменту; доступністю субстрату та кофермента; зміною каталітичної активності молекули фермента.Перший шлях регуляції є механізмом довготривалої адаптації ферментів. Для його включення і повної реалізації необхідно декілька годин або діб. Він полягає у зміні впливу на систему ядерного геному або рибосомального білкового синтезу (вплив на процеси транскрипції та трансляції) метаболітів, гормонів та інших біологічно активних речовин. Цей тип регуляції характерний здебільшого для мікроорганізмів, ферменти яких поділяють на два класи: конститутивні, які синтезуються мікроорганізмами постійно, не залежно від умов існування та адаптивні, інтенсивність біосинтезу яких змінюється залежно від змін умов існування. Адаптивні ферменти мікроорганізмів поділяють на індуцибельні та репресибельні, тобто такі, активність синтезу яких підвищується або гальмується залежно від дії певних сполук-ефекторів. Другий шлях регуляції здійснюється на рівні двох параметрів: субстрату та кофермента. Чим більша концентрація субстрату (особливо першого, вихідного), тим вища швидкість метаболічного шляху. Стосовно кофермента слід зазначити, що важливе значення для регуляції має наявність регенерованих коферментів. Наприклад, у реакціях дегідрування коферментами дегідрогеназ слугують окиснені форми НАД+, ФАД, ФМН, які відновлюються в ході реакції. Для того, щоб коферменти могли знову брати участь у реакції, необхідна їх регенерація, тобто перехід в окиснену форму. Третій шлях регуляції активності ферментів може відбуватися за чотирма основними механізмами (L. Stryеr, 1995): ковалентною модифікацією; обмеженим протеолізом; білок-білковими взаємодіями; алостерично. До регулюючих механізмів можна віднести і явище компартменталізації – строга локалізація ферментів у різних органелах, що дозволяє одночасно перебігати різноспрямованим процесам (наприклад, синтез і розпад) у межах однієї клітини. Так, процес синтезу жирних кислот відбувається в цитоплазмі, а їх розпад зосереджений у мітохондріях.
Ензимодіагностика (визначення). Зміни активності ферментів плазми та сироватки крові як діагностичні (маркерні) показники розвитку патологічних процесів (інфаркту міокарда, захворювання печінки, підшлункової залози, м’язової тканини). Ензимодіагностика полягає у постановці діагнозу захворювання (або синдрому) на базі визначення активності ферментів у біологічних рідинах людини. Для ранньої діагностики низки захворювань використання ферментів виявилося інформативнішим порівняно з іншими біохімічними тестами. Так, зміну активності аланінамінотрансферази, аспартатамінотрансферази, альдолази при інфекційному гепатиті у більшості хворих виявляють значно раніше, ніж інші біохімічні показники (тимолова проба, вміст білірубіну, білкових фракцій тощо). Підвищення активності лужної фосфатази при рахіті, креатинфосфокінази, аспартатамінотрансферази — при інфаркті міокарда використовують для ранньоїдіагностики цих захворювань. При багатьох захворюваннях зміна активності ферментів може бути настільки специфічною, що є одним із вирішальних критеріїв під час установлення діагнозу. Переконливим прикладом може бути використання сорбітолдегідрогенази для діагностики печінкових і обтураційних жовтяниць, креатинфосфокінази та аспартатамінотрансферази — для диференціації інфаркту міокарда й стенокардії. Нерідко активність ферментів змінюється ще до прояву клінічних ознак загострення. Наприклад, підвищення активності аланінамінотрансферази передує збільшенню вмісту білірубіну, погіршенню самопочуття хворого. Це допомагає своєчасно розпізнати ускладнення і змінити терапевтичну тактику. Ферменти успішно використовують у клінічній практиці для оцінювання ефективності лікування, прогнозу захворювання. Відсутність зміни активності ферментів на тлі використання лікарських та інших методів лікування свідчить про низьку їх ефективність. Так, визначення активності амінотрансфераз у сироватці крові достовірніше відображує ступінь репаративних процесів у печінці при гепатиті порівняно із вмістом білірубіну. Багато ферментів використовують у клініці для прогнозування перебігу захворювання. Наприклад, стійке зниження активності холінестерази при хронічному гепатиті свідчить про прогресування процесу й ускладнення захворювання. Різке зниження активності амінотрансфераз на тлі зростання вмісту білірубіну (ферментно-білірубінова дисоціація) свідчить про виснаження тканинних джерел ферментів за рахунок тяжкого ушкодження паренхіми печінки, що визначає відповідний прогноз. Перспективним для ферментодіагностики є дослідження ізоферментів. Доведено, що в разі ушкодження тканин ізоферменти надходять у кров та інші біологічні рідини і їхній ізоферментний спектр стає близьким до тканинного, що покладено в основу використання ізоферментів у діагностиці. Знаючи топографію ізоферментів у клітині, особливості їх тканинних і сироваткових спектрів, можна встановити локалізацію патологічного процесу. Дослідження ізоферментів має переваги перед визначенням загальної активності ферментів, оскільки їм властиві одночасно висока чутливість і специфічність. Ізоферментні спектри широко використовують для діагностики різних видів патології, насамперед у гепатології, кардіології, при захворюваннях нирок, підшлункової залози, легенів, скелетних м’язів, в онкології, гематології тощо
Ензимопатологія (визначення). Вроджені (спадкові) та набуті вади метаболізму, (приклади, їх клініко-лабораторна діагностика). Первинні, або спадкові, ензимопатії виникають унаслідок змін у генетичному коді синтезу ферментів. Причинами ферментативних дефектів можуть бути: аномальна структура ДНК, порушення перенесення генетичного коду від ДНК до РНК, змінена структура РНК і порушення в передачі інформації від РНК до рибосом. Крім того, причиною метаболічних розладів можуть бути генетично зумовлені порушення співвідношення природних активаторів та інгібіторів ферментів. Причиною спадкових ензимопатій є мутації, що виявляються характерними змінами в активності відповідних ферментів. При цьому ферментативна активність відсутня або знижена, або (дуже рідко) підвищена. Можуть з’являтися патологічні ферменти, які в нормі не трапляються.1. Галактоземії (дефіцит галактозо- 1-фосфатуридилтрансферази, або галактокінази). При цій патології відбувається накопичення в крові й тканинах галактозо-1-фосфату, вільної галактози та спирту дульциту продукту відновлення галактози. Високий їх уміст діє токсично, у немовлят після споживання молока спостерігають блювання й пронос, збільшується печінка, розвивається катаракта, затримується розумовий розвиток.2. Фруктоземії (дефіцит фруктозодифосфатальдолази, або фруктокінази). Генетичний дефект альдолази фруктозо-1-фосфату зумовлює істотні порушення в обміні вуглеводів, гіпоглікемію, ураження печінки. Для ранньої діагностики низки захворювань дослідження активності тих чи інших ферментів є значно інформативнішим порівняно з іншими біохімічними тестами. При багатьох захворюваннях зміна активності ферментів може бути настільки специфічна, що є одним із вирішальних критеріїв при встановленні діагнозу. Так, для диференційного діагнозу різних за генезом жовтяниць визначають кілька органоспецифічних ферментів (або ізоферментів) печінки, наприклад, фруктозомонофосфатальдолазу, сорбітолдегідрогеназу, для оцінки переходу гострого гепатиту в хронічний використовують АлАТ, АсАТ, аденозиндезаміназу, 5-нуклеотидазу тощо. Переконливим прикладом може служити використання креатинфосфокінази та аспартатамінотрансферази – для диференціації інфаркту міокарда й стенокардії. При захворюваннях нирок і сечовидільної системи важливе діагностичне значення має визначення активності сироваткової гліцинамідинотрансферази, ферментів сечі – лейцинамінопептидази, β-глюкуронідази, арилсульфатази.Ферменти досить чітко відображають перебіг захворювання і зарекомендували себе надійними критеріями, які характеризують період хвороби (гострий, хронічний) та її можливе загострення. Нерідко активність ферментів змінюється ще до появи характерних клінічних ознак загострення. Наприклад, підвищення активності аланінамінотрансферази передує збільшенню вмісту білірубіну, погіршенню самопочуття хворого. Це допомагає своєчасно розпізнати ускладнення і змінити терапевтичну тактику. Ферменти успішно використовують у клінічній практиці для оцінки ефективності лікування. Відсутність зміни активності ферментів на тлі використання лікарських та інших методів лікування свідчить про малу їх ефективність. Так, визначення активності амінотрансфераз в сироватці крові значно достовірніше відображає ступінь репаративних процесів у печінці при гепатиті порівняно з вмістом білірубіну. Багато ферментів використовують у клініці для прогнозування перебігу захворювання. Наприклад, стійке зниження активності холінестерази при хронічному гепатиті свідчить про прогресування процесу й ускладнення захворювання. Різке зниження активності амінотрансфераз на тлі зростання вмісту білірубіну (ферментнобілірубінова дисоціація) свідчить про виснаження тканинних джерел ферментів за рахунок тяжкого пошкодження паренхіми печінки, що визначає відповідний прогноз. Перспективним для ферментодіагностики є дослідження ізоферментів. Відомо, що при пошкодженні тканин ізоферменти надходять у кров та інші біологічні рідини й їх ізоферментний спектр стає близьким до тканинного, що лежить в основі використання ізоферментів у діагностиці. Ізоферментативні спектри широко використовують для діагностики різних видів патології. Методи прижиттєвої пункційної біопсії дають можливість досліджувати ізоферменти в тканинах людини, що достовірніше відображає картину ферментативних порушень в ураженому органі. Так, ізоферменти ЛДГ використовують для діагностики раку молочної залози, пухлин матки, передміхурової залози, мозку, шлунка, легенів, нирок тощо.
Ензимотерапія (визначення). Використання ферментів, кофакторів та інгібіторів ферментів (ацетилсаліцилова кислота, алопуринол, контрикал, сульфаніламідні препарати та інші) в якості лікарських засобів. Ензимотерапія — використання ферментів як лікарських засобів проводиться переважно в разі нестачі в організмі якогось ферменту, коферменту або як допоміжний засіб при деяких захворюваннях. Засоби замісної терапії використовують досить давно. Передусім це ферменти шлункового соку (пепсин, абомін) та підшлункової залози (панкреатин), а також багатокомпонентні препарати, що містять у своєму складі ферменти, які чинять комплексний вплив на білки, жири, вуглеводи (фестал, панзинорм, дигестал, онотон, ктазим, комбіцин). їх застосовують для поліпшення функціонального стану травного каналу та нормалізації процесів травлення. Вобензим. Препарат є спеціально підібраною комбінацією ферментів з імуномодулювальним, протинабряковим і певною мірою фібринолітичним впливом. Він чинить загальнотерапевтичну дію при запальних процесах, обмежує патологічні прояви автоімунних продуктів обміну речовин і некротизованих тканин, розсмоктує гематоми, нормалізує проникність судинних стінок, густину крові й тим самим поліпшує мікроциркуляцію.Препарат застосовують для лікування синуситу, бронхіту, бронхопневмонії, панкреатиту, виразкового коліту, хвороби Крона, розсіяного склерозу, ІХС, ревматоїдного артриту; Аспарагіназа виявляє антилейкемічну активність. Протипухлинний ефект зумовлений здатністю аспарагінази каталізувати гідроліз амінокислоти аспарагіну, необхідної лейкозним клітинам для їх розвитку: дефіцит аспарагіну впливає на клітинні мембрани, що істотно полегшує транспорт білків і поліпептидів крізь мембрани ракових клітин. Відомо, що клітини деяких злоякісних пухлин позбавлені здатності синтезувати аспарагін з інших сполук, оскільки в них відсутня аспарагінсинтетаза. Цитохром С — фермент, що бере участь у процесах тканинного дихання. Ферум, який міститься в його простетичній групі, зворотно переходить із окисненої форми у відновлену, у зв’язку з чим препарат прискорює перебіг окисних процесів. Препарат застосовують для поліпшення тканинного дихання при асфіксії новонароджених, астматичних станах, хронічній пневмонії, серцевій недостатності, ішемічній хворобі серця, інфекційному гепатиті, старечій дегенерації сітківки тощо. Велика група лікарських засобів належить до регуляторів активності ферментів, передусім до їх інгібіторів. Необхідність у них виникає досить часто, а саме: • у разі дефіциту фізіологічних інгібіторів, які виконують важливу для організму функцію — обмеження впливу ендогенних ферментів, а інколи — його захисту від ушкоджувальної дії чужорідних ферментів, зокрема мікробного походження; • під час введення з лікувальною метою ферментів у неадекватній дозі або в разі несприйняття введеного ферменту. Так, при передозуванні тромболітичних препаратів (фібринолізину), активаторів плазміну (стрептокінази, урокінази) застосовують інгібітори протеолізу (трасилол, амінокапронову кислоту тощо); • під час захворювань, у патогенезі яких певну роль відіграє гіперфункція ферментів, пов’язана з неадекватною їх активацією, аномальним викидом у кров і тканини (механічні, термічні й хімічні травми, інфекційна патологія, тромбози та емболії тощо); • під час змін ферментного спектра, при патологічному переважанні однієї ізоформи ферменту над іншою. У клінічній практиці з цією метою інгібітори широко використовують в онкології, оскільки пригнічення активності ферментів пухлинних клітин — один із відомих напрямів створення лікарських препаратів для терапії онкопатології; • у разі потреби викликати необхідну, найчастіше нефізіологічну, реакцію. На цьому принципі ґрунтується дія деяких регуляторів судинного тонусу (інгібіторів тих ферментів, які беруть участь в утворенні ангіотензину II або катехоламінів), активаторів метаболічних процесів у печінці, лікарських засобів, які пригнічують синтез простагландинів тощо.Інгібітори ферментів, потенційно придатні для застосування в терапії, досить поширені в природі, їх також можна отримувати шляхом синтезу. Більшість інгібіторів тваринного й рослинного походження, вивчені в експерименті чи клініці, є поліпептидами з молекулярною масою понад 5 000, тоді як мікробні інгібітори, як правило, мають невелику молекулярну масу. Інгібітори, виділені з рослин і мікроорганізмів, належать переважно до простих білків, а інгібітори тваринного походження часто містять у своєму складі вуглеводи. Наприклад, значна кількість інгібіторів протеаз тваринного походження є глікопротеїнами. Для деяких рослинних інгібіторів характерний низький уміст ароматичних амінокислот. Слід зазначити, що багато мікроорганізмів продукують хімічні сполуки, здатні впливати на ферментативні процеси в тканинах організмів тварин і людини.
Механізм дії ферментів – фармацевтичних препаратів при захворюваннях травної системи, при гнійно-некротичних процесах, як фібринолітичних препаратів та інше. У якості лікарських препаратів-антиметаболітів використовують аналоги нуклеотидів для лікування онкологічних захворювань, сульфаніламідні препарати (аналоги параамінобензойної кислоти (ПАБК)) для лікування інфекційних захворювань. ПАБК структурно подібна до сульфанілової кислоти, похідні якої є сульфаніламідними препаратами. У мікроорганізмах в присутності ПАБК синтезується фолієва кислота, яка є важливим коферментом низки ферментів, що беруть участь у синтезі нуклеїнових кислот, а отже, і білків, тобто фолієва кислота є фактором росту бактерій, зокрема, стафілококів, пневмококів тощо. Цим забезпечується ріст і розмноження мікроорганізмів. У фолієвій кислоті ПАБК має два замісники: гетероциклічне похідне (R1) і глутамінову кислоту (R2). Сульфаніламідні препарати конкурують з ПАБК (структурна подібність) на стадії утворення фолієвої кислоти. Наявність сульфамідної групи в сульфаніламідних препаратах перешкоджає взаємодії ПАБК з глутаміновою кислотою, цим самим припиняється (блокується) біосинтез фолієвої кислоти. Гальмування синтезу фолієвої кислоти в мікроорганізмах призводить до порушення біосинтезу нуклеїнових кислот і білків, внаслідок цього пригнічується ріст і розмноження бактерій.Таким чином, сульфанілова кислота та її N-заміщені аміди, виступають антиметаболітами ПАБК, зумовлюють бактеріостатичний ефект.


Активатори та інгібітори ферментів: приклади та механізми дії. Активування ферментів. Речовини, які підвищують активність ферментів, одержали назву активаторів. Присутність активатора вкрай важлива для ферменту. До активаторів належать кофактори, іони металів, різноманітні модифікатори тощо. Субстрат у певних межах концентрацій є активатором - після досягнення насичених концентрацій субстрату активність ферменту не зростає. Субстрат полегшує формування потрібної конформації активного центру ферменту (індукція), підвищує його стабільність. Досить часто роль активаторів виконують іони металів: вони можуть входити до складу каталітичної ділянки активного центру ферменту; сприяти зв'язуванню субстрату з посадочною ділянкою ферменту (зв'язуючий місток); іноді метали можуть сполучатися не з ферментом, а із субстратом, утворюючи металосубстратний комплекс, на який краще діє фермент; вони можуть діяти непрямим шляхом, зв'язуючи присутній інгібітор тощо. Активація деяких ферментів (особливо тих, що виробляються в шлунково-кишковому тракті) може відбуватися протеолітичним шляхом. Гальмування ферментів. Дія багатьох ферментів може бути загальмована, а в ряді випадків і повністю припинена під час дії певних хімічних речовин - інгібіторів. Інгібітори з тієї чи іншої причини частково або повністю перешкоджають утворенню продуктивного фермент-субстратного комплексу.
Типи
інгібірування ферментів: зворотне
(конкурентне, неконкурентне) та незворотнє
інгібування.
Конкурентні інгібітори за будовою
подібні до субстрату, вони конкурують
із ним за зв’язування з активним центром
фермента (рис. 1.13). Характерною особливістю
конкурентного гальмування є те, що
ефективність інгібітора залежить від
співвідношення концентрацій субстрату
й інгібітора. При наявності субстрату
[S] та інгібітора [І] одночасно відбуваться
дві реакції: Неконкурентним
інгібуванням
називають
таке гальмування, при якому інгібітор
не є структурним аналогом субстрату,
він взаємодіє з ферментом або в ділянці,
відмінній від активного центра, або з
уже сформованим фермент-субстратним
комплексом. Приєднання неконкурентного
інгібітора викликає зміну конформацію
молекули фермента в такий спосіб, що
порушується взаємодія субстрату з
активним центром фермента, що призводить
до зниження швидкості реакції (рис.
1.16). У випадку утворення потрійного
комплексу (фермент-субстрат-інгібітор)
останній не спроможний перетворитися
на продукт, у результаті чого реакція
зупиняється.
Неконкурентним
інгібуванням
називають
таке гальмування, при якому інгібітор
не є структурним аналогом субстрату,
він взаємодіє з ферментом або в ділянці,
відмінній від активного центра, або з
уже сформованим фермент-субстратним
комплексом. Приєднання неконкурентного
інгібітора викликає зміну конформацію
молекули фермента в такий спосіб, що
порушується взаємодія субстрату з
активним центром фермента, що призводить
до зниження швидкості реакції (рис.
1.16). У випадку утворення потрійного
комплексу (фермент-субстрат-інгібітор)
останній не спроможний перетворитися
на продукт, у результаті чого реакція
зупиняється. Неконкурентні
інгібітори можуть бути проміжними
продуктами метаболізму, які утворюються
в живих організмах. Вони здатні зменшувати
швидкість реакції (Vмакс),
але не впливати на спорідненість
ферменту до субстрату (Км)
(рис. 1.17).Неконкурентними інгібіторами
є ціаніди, які міцно сполучаються з
тривалентним залізом, яке входить в
каталітичну ділянку гемінового фермента
цитохромоксидази. Блокування останнього
призводить до припинення тканинного
дихання та загибелі клітини.Велика
токсичність
іонів
ртуті, свинцю, миш'яку обумовлена їх
властивістю блокувати SH-групи каталітичних
ділянок низки ферментів. Проте, важкі
метали лише в невеликих концентраціях
виконують роль неконкурентних
інгібіторів, у великих кількостях вони
є інактиваторами і діють як денатуруючі
агенти. Для подолання інтоксикації,
викликаної цими препаратами, застосовують
різні реактиватори (або протиотрути),
які витісняють інгібітор з комплексу.
До них відносять, наприклад, SH-вмісні
комплекси (цистеїн, унітіол тощо),
лимонну кислоту, етилендіамінтетраацетатну
кислоту тощо. Безконкурентне
інгібування.
Цей
тип інгібування
спостерігають
у тому випадку, коли інгібітор зворотно
взаємодіє з ферментом тільки після
утворення фермент-субстратного
комплексу, тобто безконкурентний
інгібітор не сполучається з ферментом
у відсутності субстрату. Інгібітор
полегшує приєднання субстрату, а потім,
зв'язуючись, інгібує фермент. Це рідкісний
вид інгібування. Субстратне
інгібування. Наявність
надмірно високої концентрації субстрату
викликає гальмування ферментативної
реакції. Пояснюється цей факт тим, що
молекули субстрату в надлишку займають
неправильне положення в активному
центрі фермента.
Неконкурентні
інгібітори можуть бути проміжними
продуктами метаболізму, які утворюються
в живих організмах. Вони здатні зменшувати
швидкість реакції (Vмакс),
але не впливати на спорідненість
ферменту до субстрату (Км)
(рис. 1.17).Неконкурентними інгібіторами
є ціаніди, які міцно сполучаються з
тривалентним залізом, яке входить в
каталітичну ділянку гемінового фермента
цитохромоксидази. Блокування останнього
призводить до припинення тканинного
дихання та загибелі клітини.Велика
токсичність
іонів
ртуті, свинцю, миш'яку обумовлена їх
властивістю блокувати SH-групи каталітичних
ділянок низки ферментів. Проте, важкі
метали лише в невеликих концентраціях
виконують роль неконкурентних
інгібіторів, у великих кількостях вони
є інактиваторами і діють як денатуруючі
агенти. Для подолання інтоксикації,
викликаної цими препаратами, застосовують
різні реактиватори (або протиотрути),
які витісняють інгібітор з комплексу.
До них відносять, наприклад, SH-вмісні
комплекси (цистеїн, унітіол тощо),
лимонну кислоту, етилендіамінтетраацетатну
кислоту тощо. Безконкурентне
інгібування.
Цей
тип інгібування
спостерігають
у тому випадку, коли інгібітор зворотно
взаємодіє з ферментом тільки після
утворення фермент-субстратного
комплексу, тобто безконкурентний
інгібітор не сполучається з ферментом
у відсутності субстрату. Інгібітор
полегшує приєднання субстрату, а потім,
зв'язуючись, інгібує фермент. Це рідкісний
вид інгібування. Субстратне
інгібування. Наявність
надмірно високої концентрації субстрату
викликає гальмування ферментативної
реакції. Пояснюється цей факт тим, що
молекули субстрату в надлишку займають
неправильне положення в активному
центрі фермента.
Регуляція ферментативних процесів. Шляхи та механізми регуляції: алостеричні ферменти; ковалентна модифікація ферментів, обмежений протеоліз. Дія регуляторних білків. Зазвичай кожен метаболічний шлях має свої ключові ферменти, які називають регуляторними, оскільки завдяки їм відбувається регуляція швидкості всього шляху. Ці ферменти можуть каталізувати початкові, або незворотні або найповільніші реакції, вони також розташовуються в точках розгалуження метаболічного шляху. Впливаючи на такі ферменти модифікаторами (активаторами чи інгібіторами) можна змінити швидкість перебігу не лише однієї реакції, а й усього метаболічного шляху.Будь-які зміни оточуючого чи внутрішнього середовища вимагають включення адаптаційних процесів, що реалізуються, першою чергою, через зміну швидкості тої чи іншої ферментативної реакції.Регуляція швидкості ферментативної реакції здійснюється трьомах шляхами: зміною кількості ферменту; доступністю субстрату та кофермента; зміною каталітичної активності молекули фермента.Перший шлях регуляції є механізмом довготривалої адаптації ферментів. Для його включення і повної реалізації необхідно декілька годин або діб. Він полягає у зміні впливу на систему ядерного геному або рибосомального білкового синтезу (вплив на процеси транскрипції та трансляції) метаболітів, гормонів та інших біологічно активних речовин. в цитоплазмі, а їх розпад зосереджений у мітохондріях. Ковалентна модифікація ферментів – один із механізмів контролю метаболічних процесів, що може відбуватися шляхом зворотного фосфорилування-дефосфорилування, метилування, аденілування, АДФ-рибозилування білків-ферментів. Фосфорилують білки спеціальні ферменти протеїнкінази, які за допомогою залишку фосфату АТФ здійснюють фосфорилування серинового, тирозинового чи треонінового радикалу відповідного білка. Зворотну реакцію – дефосфорилування білків – каталізують протеїнфосфатазами. Активація ферментів шляхом обмеженого протеолізу. Активація низки ферментів (здебільшого травного тракту або плазми крові) може відбуватися протеолітичним шляхом. Такі ферменти синтезуються в неактивній формі у вигляді проферментів або зимогенів (попередників ферментів), активний центр яких замаскований додатковою ділянкою пептидного ланцюга, внаслідок чого субстрат не може взаємодіяти з активним центром. Активація шляхом білок-білкових взаємодій включає в себе два механізми: активацію в результаті приєднання регуляторних білків і зміну каталітичної активності внаслідок асоціації чи дисоціації протомерів фермента. До регуляторних білків, які можуть чинити вплив на активність ферментів належать: кальмодулін-кальційвмісний білок, який, зв'язуючись з чотирма іонами кальцію, сприяє збільшенню цитозольної концентрації останнього при певних біохімічних та фізіологічних процесах у клітині. Алостерична регуляція ферментів. Активування ферментів може здійснюватися шляхом приєднання до алостеричного центра фермента специфічної модифікуючої групи (активатора), що сприяє зміні конформації фермента, його активного центра та, як наслідок, прискоренню ферментативної реакції (алостерична активація). Вона, основним чином, характерна для олігомерних ферментів, які складаються з кількох протомерів або мають доменну структуру.
Циклічні
нуклеотиди (цАМФ, цГМФ) як регулятори
ферментативних реакцій та біологічних
функцій клітини. Важливою
та поширеною біологічною системою
контролю за ферментативними реакціями,
що поєднує в собі різні молекулярні
механізми регуляції, є система циклічних
нуклеотидів. Циклічні нуклеотиди
3',5'-АМФ (цАМФ) та 3',5' ГМФ (цГМФ) - це
внутрішні (3'5' дифосфорні ефіри аденілової
(АМФ) та гуанілової (ГМФ) кислот. Найбільш
поширеними є цАМФ-залежні системи
контролю за внутрішньоклітинними
біохімічними процесами, зокрема за
такими, що підлягають нейрогуморальній
регуляції з боку цілісного організму,
яка реалізується гормонами та
нейромедіаторами. Регуляція ферментативних
процесів за участю цАМФ включає декілька
послідовних
стадій
передавання і трансформації хімічного
(регуляторного) сигналу.
1.
Утворення циклічних нуклеотидів у
реакціях, що каталізуються ферментами
циклазами: аденілатциклазою та
гуанілатциклазою з нуклеозидтрифосфатів
АТФ та ГТФ, відповідно:
Розщеплення
цАМФ та цГМФ до звичайних, нециклічних
нуклеозидмонофосфатів каталізується
фосфодіестеразою циклічних нуклеотидів.
Фермент аденілатциклаза розміщений у
плазматичних мембранах клітин і його
активація відбувається в результаті
взаємодії з рецепторами мембран певних
фізіологічно активних сполук, зокрема
гормонів адреналіну, глюкагону тощо.
2.
Активація циклічним АМФ протеїнкіназ,
функцією яких є фосфорилування інших
ферментних білків. Ці
цАМФ-залежні протеїнкінази є регуляторними
ферментами, що активуються цАМФ за
механізмом алостеричного контролю.

Принципи
та методи виявлення ферментів у
біооб'єктах. Одиниці виміру активності
та кількості ферментів. Оскільки
кількість ферменту в біологічному
об'єкті у більшості випадків визначити
неможливо, для характеристики швидкості
біохімічної реакції, що каталізується
певним ферментом, за умов сталості
інших показників середовища
(фізико-хімічних параметрів, концентрації
активаторів та інгібіторів) користуються
значеннями активності ферменту. Активність
ферменту -
це умовна величина, що прямо пропорційна
швидкості біохімічної реакції, яку
каталізує певний фермент. У свою чергу,
як легко зрозуміти, швидкість
ферментативної реакції можна визначити
або за кількістю субстрату (S), що
перетворився за певний проміжок часу,
або за кількістю накопиченого за цей
час продукту реакції (Р). У біохімічній
практиці для кількісної характеристики
реакцій, що каталізуються ферментами,
використовують умовні величини - одиниці
ферменту. Загальновживаними є такі
одиниці ферменту:
1.
За одиницю ферменту U (unit, англ.), що
рекомендована Міжнародним біохімічним
союзом (МБС), приймають таку його
кількість, яка каталізує перетворення
1 мкмоль субстрату за 1 хвилину: ![]() 2.
При використанні одиниць системи СІ
(SI) активність ферменту виражають у
каталах (кат). 1 Катал (кат) - така кількість
ферменту, яка каталізує перетворення
1 моля субстрату за 1 с:
2.
При використанні одиниць системи СІ
(SI) активність ферменту виражають у
каталах (кат). 1 Катал (кат) - така кількість
ферменту, яка каталізує перетворення
1 моля субстрату за 1 с: ![]() 3.
Розповсюдженою одиницею є питома
активність ферменту, яка визначається
кількістю одиниць ферментної активності,
що припадають на 1 мг білка в біологічному
об'єкті (U/мг білка). У медичній ензимології
активність ферменту часто виражають
в одиницях (U) на 1 л досліджуваної
біологічної рідини - сироватки крові,
слини, сечі тощо (U/л).
3.
Розповсюдженою одиницею є питома
активність ферменту, яка визначається
кількістю одиниць ферментної активності,
що припадають на 1 мг білка в біологічному
об'єкті (U/мг білка). У медичній ензимології
активність ферменту часто виражають
в одиницях (U) на 1 л досліджуваної
біологічної рідини - сироватки крові,
слини, сечі тощо (U/л).
Поняття про обмін речовин та енергії. Характеристика катаболічних, анаболічних та амфіболічних шляхів метаболізму, їх значення. . Метаболізм, що є найбільш характерною ознакою та неодмінною умовою існування будь-якої біологічної системи, поділяється на анаболізм (асиміляцію, синтез) та катаболізм (дисиміляцію, розщеплення молекул).Катаболізм. Процеси катаболізму являють собою сукупність реакцій розщеплення хімічних сполук, що надходять в організм у вигляді продуктів харчування та містять чужорідні вуглеводи, ліпіди, білки тощо. Крім того, в будь-якій клітині постійно відбувається розщеплення власних біомолекул. Катаболізм є екзергонічним процесом, тобто таким, що призводить до вивільнення хімічної енергії, яка частково використовується організмом в ході анаболізму. Ця хімічна енергія звільняється в результаті реакцій окислення біомолекул - проміжних продуктів внутрішньоклітинного розщеплення моносахаридів: переважно глюкози, жирних кислот, гліцерину та деяких амінокислот. Основні реакції біологічного окислення, що вивільняють енергію, необхідну для процесів життєдіяльності, відбуваються в мітохондріях (саркосомах), у мембранах яких локалізовані також складні ферментні та йонтранспортуючі системи, які реалізують накопичення енергії окислювальних процесів у вигляді високоенергетичних (макроергічних) зв'язків АТФ.Анаболізм являє собою постійний синтез молекул та побудову структур власного організму з органічних сполук, що надходять з навколишнього середовища у вигляді продуктів харчування та продуктів їх перетворення (інтермедіатів). Процеси анаболізму є ендергонічними, тобто такими, що потребують витрат хімічної енергії, яка постачається за рахунок реакцій катаболізму, переважно у формі молекул АТФ. Таким чином, живі організми за термодинамічними закономірностями їх функціонування є не тепловими машинами, а хімічними машинами, тобто системами, в яких різні види роботи здійснюються за рахунок хімічної енергії молекул, до того ж при сталій температурі (у людини близько 37 °С). При цьому джерелом хімічної енергії для ендергонічних процесів синтезу та побудови нових клітинних структур є екзергонічні реакції біологічного окислення, що відбуваються в електротранспортних ланцюгах мітохондрій та акумулюють цю енергію в ході окисного фосфорилування.
Екзергонічні та ендергонічні біохімічні реакції; роль АТФ та інших макроергічних фосфатів у їх спряженні. Обмін речовин в організмі безпосередньо пов’язаний з обміном енергії. Серед реакційметаболізму розрізняють екзергонічні та ендергонічні реакції. Екзергонічні реакції (процеси) — такі, що супроводжуються вивільненням хімічноїенергії, необхідноїдляфункціонуванняживихорганізмів. Найбільш важливими екзергонічними процесами вживих системах є окислю- вальні реакції, що каталізуються окислювально-відновлювальними ферментами мембран мітохондрій. Енергія, яка вивільняється в окислювальних реакціях, здебільшого акумулюється у формі макроергічних сполук. Ендергонічні реакції (процеси) — такі, що потребують для своєї течії витрат енергії; цеферментативні реакції синтезу та відновлення: – реакції синтезупризводять до утворення нових хімічних зв’язків, ускладнення структурибіомолекул (простихсполук табіополімерів); заджерелоенергіївбільшості реакцій синтезу правлятьмакроергічнізв’язки АТФ;. Спряження ендергонічних процесів з екзергонічними здійснюється за ра- хунок синтезу протягом екзергонічної реакції сполук з високим енергетич- ним потенціалом, які в подальшому використовуються в ендергонічних реакціях, що забезпечує передачу хімічної енергії від екзергонічного до ендер- гонічного процесу. Головною макроергічною сполукою в живих організмах є молекула АТФ. У структурі молекули АТФ є два макроергічні зв’язки, молекули АДФ — один макроергічний зв’язок: До макроергічних сполук належать також такі інтермедіати вуглеводного, ліпідного і амінокислотного метаболізму, як фосфо- енолпіруват, 1,3-дифосфогліцерат та важливий енергетичний резерв м’язів — креатинфосфат.
Внутрішньоклітинна локалізація метаболічних шляхів, компартменталізація метаболічних процесів в клітині. Виділення субклітинних структур методом диференційного центрифугування. Окремі метаболічні шляхи містяться у визначених внутрішньоклітинних ділянках — компартментах, що відокремлені біологічними мембранами, зокрема: – в ядрах — реакції біосинтезу (реплікації) ДНК, синтезу інформаційних та інших класів РНК (транскрипції); – в мітохондріях — реакції циклу трикарбонових кислот, βββββ-окислення жирних кислот, електронного транспорту та окисного фосфорилювання; – в цитоплазмі (цитозолі) — реакції гліколізу, глюконеогенезу, синтезу жирних кислот, обміну амінокислот тощо; – в рибосомах — реакції білкового синтезу (утворення поліпептидних ланцюгів); – у мембранах ендоплазматичного ретикулуму — цитохром Р-45О-залежні процеси окисного гідроксилювання гідрофобних субстратів ендогенного та екзогенного походження (реакції “мікросомального окислення”); – у лізосомах — реакції гідролітичного розщеплення біополімерів — білків та нуклеїнових кислот (як власних біомолекул, так і чужорідних сполук, що поглинаються певними клітинами за рахунок ендоцитозу). Для вивчення біохімічних реакцій та окремих метаболітів, які локалізовані в певних клітинних компартментах, використовують метод диференційного центрифугування. Сутність методу полягає в послідовному центрифугуванні з різними швидкостями гомогенатів біологічних тканин (печінки, м’язів, мозку тощо), що містять у собі суспендовані субклітинні структури (ядра, мітохондрії, лізосоми, різні мембранні везикули, рибосоми). Для отримання гомогенату тканину дезінтегрують у спеціальному гомогенізаторі, суспендуючи її в біологічно інертному розчині (О,25 М розчин сахарози, сольові розчини). Залежно від розмірів та маси, означені органели осідають (седиментують) з різною швидкістю, тому, застосовуючи різну швидкість обертання ротору ультрацентрифуги (та, відповідно, збільшуючи фактор седиментації, що кількісно визначається в одиницях прискорення земного тяжіння — g) досягають фракціонування тканинного гомогенату на окремі субклітинні структури
Етапи катаболізму біомолекул: білків, вуглеводів, ліпідів; їх характеристика. У ферментативному розщепленні складних біоорганічних сполук в організмі виділяють три основних стадії (етапи), що є загальними для катаболізму різних біомолекул. Стадія 1. На першій стадії катаболізму складні молекули (макромолекули вуглеводів, білків, нуклеїнових кислот та молекули ліпідів) розщеплюються до простих компонентів: – полісахариди — до моносахаридів (переважно — глюкози, фруктози, галактози); – ліпіди (триацилгліцероли) — до жирних кислот та гліцеролу; – білки — до амінокислот; – нуклеїнові кислоти — до нуклеотидів. Реакції першої стадії катаболізму є гідролітичними за своїм механізмом і каталізуються гідролазами травного тракту (шлунка, кишечника). Ці реакції не супроводжуються суттєвим вивільненням хімічної енергії. Стадія 2. На другій стадії катаболізму декілька десятків метаболітів, що утворились на першій стадії, підлягають ферментативним реакціям розщеплення з вивільнення певної кількості хімічної енергії, яка акумулюється у високо-енергетичних (макроергічних) зв’язках АТФ. Реакції другої стадії катаболізму відбуваються внутрішньоклітинно (в цитоплазмі та частково в мітохондріях). Основними з цих реакцій є: для моносахаридів — гліколіз, кінцевим метаболітом якого є піровиноградна кислота (піруват), що в подальшому окислюється до активної форми оцтової кислоти — ацетил-коензиму А (ацетил-КоА); для жирних кислот — βββββ-окислення, кінцевим продуктом якого є ацетил-КоА; для гліцеролу — розщеплення до пірувату, який перетворюється в ацетил-КоА; для амінокислот та нуклеотидів — дезамінування з виділенням аміаку та розщепленням безазотистих молекулярних скелетів до дво- і тривуглецевих карбонових кислот та їх похідних; більшість із цих метаболітів у кінцевому підсумку також утворюють ацетильний радикал у формі ацетил-КоА. Таким чином, ацетил-КоА — це загальний кінцевий продукт другої стадії внутрішньоклітинного катаболізму вуглеводів, ліпідів та амінокислот. Стадія 3. На третій стадії катаболізму відбувається окислення ацетил-КоА до кінцевих метаболітів — двоокису вуглецю та води. Ця стадія має місце в мітохондріях і складається з двох процесів: – циклу трикарбонових кислот (ЦТК, циклу Кребса), в результаті функціонування якого утворюється СО2, а атоми водню використовуються для відновлення коферментів нікотинамідаденіндинуклеотиду (НАД+) та флавінаденінди нуклеотиду (ФАД); – системи електронного транспорту в мембранах мітохондрій, в якій атоми водню (протони та електрони) переносяться на кисень з утворенням Н2О; реакцій біологічного окислення використовується для синтезу молекул АТФ — головного безпосереднього постачальника енергії у всіх біологічних ендергонічних процесах.
Найважливіші метаболіти шляхів обміну білків, вуглеводів, ліпідів; їх роль в інтеграції метаболізму клітини. Серії реакцій в клітині переважно об'єднуються у метаболічні шляхи, в яких продукт одного перетворення стає субстратом для наступного. Зазвичай ферменти одного метаболічного шляху групуються разом, утворюючи великі мультиензимні комплекси. Деякі шляхи забезпечують розщеплення складних органічних речовин і запасання енергії у формі хімічних зв'язків АТФ, в результаті чого його концентрація в клітині підтримується на досить високому рівні. Також в цих реакція відновлюються НАД і НАДФ. Сукупність таких метаболічних шляхів називається катаболізмом (дисиміляція, енергетичний обмін). Інші шляхи полягають у синтезі складних молекул із простіших попередників (наприклад полімерів із мономерів), вони завжди потребують енергії і описуються загальним терміном анаболізм (асиміляція, пластичний обмін). Зв'язними ланками між катаболізмом та анаболізмом є АТФ та інші нуклеотидтрифосфати. Інколи виділяють ще так звані амфіоблічні шляхи — перехрестя катаболізму та анаболізму. Хоча уявлення про метаболізм як сукупність окремих шляхів зручне для його вивчення і систематизації, воно надто спрощене, щоб відповідати реальності. Багато із проміжних продуктів є частиною кількох шляхів. Через це метаболізм найкраще описується як складна сітка взаємопов'язаних реакцій, в якій зміна концентрації навіть однієї речовини викликає суттєві наслідки у багатьох її частинах. забезпечується основними шляхами: активацією або пригніченням синтезу певних ферментів, та зміною їхньої активності. Перший метод повільніший і дає стійкіші результати, другий дозволяє миттєву відповідь. Багато ключових ферментів метаболічних шляхів є алостеричними, тобто такими, що крім активного центру мають додатковий сайт для зв'язування регуляторних молекул, що змінюючи конформацію білка впливають на його активність. Алостеричні модулятори можуть бути як активуючими так і інгібуючими. Окрім того, регуляція метаболічних шляхів може відбуватись шляхом зміни доступності субстратів. Наприклад, багато клітин можуть розщеплювати глюкозу тільки в тоді, коли на них діє інсулін, щостимулює транспорт цієї речовини із крові. В еукаріотичних клітинах протилежно напрямлені метаболічні шляхи часто розподілені по різних компартментах. Наприклад, окиснення жирних кислот відбувається у мітохондріях, а синтез — у цитоплазмі. Перехід субстратів із одного компартменту в інший може слугувати точкою контролю. Оскільки одним із першочергових завдань метаболізму є підтримання гомеостазу (динамічної сталості внутрішніх умов), більшість його регуляторних шляхів організовані за принципом негативного зворотного зв'язку: метаболічний шлях пригнічується його кінцевим продуктом, що діє на один із перших ферментів цього шляху. Наприклад амінокислота ізолейцин синтезується із треоніну у п'ять кроків. Коли в клітині виробляється більше ізолейцину ніж потрібно для синтезу білків, його надлишок пригнічує перший фермент цього метаболічного шляху і синтез припиняється до того часу, поки концентрація амінокислоти не зменшиться.
Цикл трикарбонових кислот (ЦТК): внутрішньоклітинна локалізація ферментів ЦТК; послідовність реакцій ЦТК; характеристика ферментів та коферментів ЦТК; реакції субстратного фосфорилування в ЦТК; значення в обміні печовин Цикл трикарбонових кислот (ЦТК) — це загальний кінцевий шлях окислювального катаболізму клітини в аеробних умовах. Реакції іферменти ЦТКлокалізовані в матриксі та внутрішній мембрані мітохондрій. Вони функціонально та біохімічно спряженізмітохондріаль- ними електронотранспортними ланцюгами, що викори- стовують для відновлення атомів кисню відновлю- вальні еквіваленти від НАДН (НАДН + Н+ ) та ФАДН2 або ФМНН2 і утворюють АТФ у ході окисного фосфорилювання. Цикл трикарбонових кислот починається з взаємо- дії (конденсації) двовуглецевої молекули ацетил-КоА (C2 ) з чотиривуглецевою (С4 ) щавлевооцтовою кис- лотою (оксалоацетатом), що призводить до утворення шестивуглецевої (С6 ) молекули лимонної кислоти (цитрату). В результаті подальшого багатоступеневого перетворення три- та дикарбоно- вих кислот (інтермедіатів ЦТК) відбувається регенерація оксало- ацетату (С4 ) та виділяються дві молекули двоокису вуглецю (С2 ).роль








Цикл
трикарбонових кислот (ЦТК): механізми
регуляції, вплив алостеричних модуляторів.
Анаплеротичні реакції ЦТК. Енергетичний
баланс циклу трикарбонових кислот.
Цикл трикарбонових кислот (ЦТК) — це
загальний кінцевий шлях окислювального
катаболізму клітини в аеробних умовах.
Реакції іферменти ЦТКлокалізовані в
матриксі та внутрішній мембрані
мітохондрій. Вони функціонально та
біохімічно спряженізмітохондріаль-
ними електронотранспортними ланцюгами,
що викори- стовують для відновлення
атомів кисню відновлю- вальні еквіваленти
від НАДН (НАДН + Н+ ) та ФАДН2 або ФМНН2 і
утворюють АТФ у ході окисного
фосфорилювання. Цикл трикарбонових
кислот починається з взаємо- дії
(конденсації) двовуглецевої молекули
ацетил-КоА (C2
) з чотиривуглецевою (С4
) щавлевооцтовою кис- лотою (оксалоацетатом),
що призводить до утворення шестивуглецевої
(С6 ) молекули лимонної кислоти (цитрату).
В результаті подальшого багатоступеневого
перетворення три- та дикарбоно- вих
кислот (інтермедіатів ЦТК) відбувається
регенерація оксало- ацетату (С4 ) та
виділяються дві молекули двоокису
вуглецю (С2 ).Біохімічний
підсумок циклу трикарбонових кислот
полягає в утворенні двох молекул СО2
(в ізоцитратдегідрогеназній та
ααααα-кетоглутаратдегідрогеназній
реакціях) та чотирьох пар атомів водню,
три з яких акцептуються НАД+ та одна —
ФАД. Відновлені коферменти окислюються
в дихальному ланцюзі мітохондрій,утворюючи
за рахунок окисного фосфорилювання по
3 молекули АТФ на кожну молекулу НАДН
і по 2 молекули АТФ на кожну молекулу
ФАДН2. Крім того, одна молекула АТФ
утворюється в субстратному фосфорилюванні
при перетворенні сукциніл-КоА в сукцинат.
Таким чином, при повному окисленні
однієї молекули ацетил-КоА до СО2 та
Н2О в циклі трикарбонових кислот
генерується 12 молекул АТФ.

Піридинзалежні дегідрогенази. Будова НАД+ і НАДФ+. Їх значення у реакціях окиснення та відновлення. Коферментами цих дегідрогеназ є нуклеотиди НАД+ або НАДФ+, у структурі молекул яких міститься похідне піридину — нікотинамід (глава 6). Зв’язок між НАД+ (або НАДФ+ ) та білковою частиною ферменту (апофер- ментом) у складі піридинзалежних дегідрогеназ нестійкий: він утворюється та руйнується в процесі каталітичного циклу, що дозволяє вважати нікотинамідні нуклеотиди скоріше субстратами, ніж простетичними групами. Реакції, що каталізуються НАД(Ф)-залежними дегідрогеназами, можуть бути зображені в загальному вигляді такими рівняннями: Активною структурою в молекулі НАД+ або НАДФ+ , що акцептує віднов- лювальні еквіваленти від субстрату, є піридинове кільце никотинаміду. У ході ферментативної реакції субстрат відщеплює два атоми водню (2Н+ + 2е– ), один з яких у формі гідрид-іону: Н– (тобто Н+ + 2е– ) приєднується до піридинового кільця НАД(Ф) + , а другий у вигляді протону (іону Н+ ) надходить у реакційне середовище: Як свідчить наведене рівняння, під час реакції до четвертого вуглецевого атома нікотинаміду приєднується атом водню (тобто Н+ + е– ), а додатковий електрон гідрид-іону взаємодіє з азотом піридинового кільця. В подальшому викладенні тексту відновлені форми нікотинамідних коферментів (системи НАД(Ф)Н + Н+ ) для спрощення будуть в деяких випадках позначатися як НАД(Ф)Н. Дегідрогенази, залежні від нікотинамідних коферментів, дуже поширені вживих клітинах. Вони виконують функції анаеробних дегідрогеназ, що відщеплюють протони та електрони від багатьох субстратів, відновлюючи НАД+ або НАДФ+ , передаючи в подальшому відновлювальні еквіваленти на інші акцептори. НАД-залежні дегідрогенази — ці ферменти каталізують окислювально- відновлювальні реакції, що містяться на окислювальних шляхах метаболізму — гліколізу, циклу лимонної кислоти, β-окислення жирних кислот, окисного дезамінування амінокислот, дихального ланцюга мітохондрій. НАДФ-залежні дегідрогенази — ці ферменти беруть участь у процесах від- новлювального синтезу, що відбуваються в цитозолі, зокрема постачають атоми водню при синтезіжирних кислот та стероїдів. Головним джерелом відновленого НАДФ є дегідрогеназні реакції пентозофосфатного шляху окислення глюкози.

Флавінзалежні дегідрогенази. Будова ФАД і ФМН. Їх роль у реакціях окиснення та відновлення. Дегідрогенази цього типу за хімічною природою єфлавопротеїнами, просте- тичними групами, в яких є флавінаденіндинуклеотид (ФАД) та флавінмоно- нуклеотид (ФАД) (будову див. главу 6). На відміну від піридинзалежних дегідрогеназ, у більшості флавінзалежних ферментів коферменти (ФАД та ФМН) міцно зв’язані з білковою частиною і не відщеплюються від неї нажодній стадії каталітичного циклу. ВиключеннямєФАД- залежна оксидаза D-амінокислот, у складі якої білок має низьку спорідненість із коферментом. Загальні рівняння окислення субстратів за участю флавінзалежних дегідро- геназ: Активною частиною молекули ФАД або ФМН, що бере участь в окислю- вально-відновлювальній реакції, є ізоалоксазинове кільце рибофлавіну, яке акцептує два атоми водню (2Н+ + 2е– ) від субстрату: У процесах біологічного окислення певні флавопротеїнові ферменти відігра- ють роль як анаеробних, так і аеробних дегідрогеназ. Флавопротеїни — анаеробні дегідрогенази: – НАДН-дегідрогеназа — ФМН-залежний компонент внутрішньої мембрани мітохондрій; виконує функцію колектора електронів, що забирає їх від НАДН і передає на більш електропозитивні компоненти дихального ланцюга мітохондрій; – сукцинатдегідрогеназа — ФАД-залежний фермент циклу трикарбонових кислот, що окислює янтарну кислоту; – дигідроліпоїлдегідрогеназа — ФАД-залежний фермент, що бере участь в окислювальному декарбоксилюванні піровиноградної кислоти; – ацил-КоА-дегідрогеназа — ФАД-залежний фермент системи β-окислення жирних кислот; – гліцерол-3-фосфат-дегідрогеназа — ФАД-залежний фермент, що окислює гліцерол-3-фосфат у мітохондріях. Флавопротеїни — аеробні дегідрогенази: – дегідрогеназа (оксидаза) L-амінокислот — ФМН-залежний фермент нирок, специфічний до природних L-амінокислот; – ксантиноксидаза (ксантиндегідрогеназа) — ФАД-залежний фермент, який окислює пурини до сечової кислоти; – глюкозоксидаза — ФАД-залежний рослинний фермент, що використовується для кількісного визначення глюкози в біологічних рідинах.

Убіхінон, будова та його роль у реакціях окиснення та відновлення. Убіхінон є останнімкомпонентомдихального ланцюгамітохондрій, що здатний транспортувати як електрони, так і протони. На рівні цитохромуb шляхи електронів і протонів розділяються — протони переходять з внутрішньої поверхнімітохондрі- альної мембрани на зовнішню, а електрони через послідовність цитохромів транспортуються на цитохромa3 , який відновлює кисень: 2 e– + 1/2 O2 O2– Взаємодія відновленого кисню з вільними протонами мітохондріального матриксу призводить до утворення молекули води: 2 Н+ + О2– Н2 O
Цитохроми та їх роль у тканинному диханні. Будова їх простетичної групи. Цитохроми — залізовмісні білки мітохондрій, що належать до класу гемо- протеїнів. У цитохромах іон заліза входить до складу металопорфіринового комплексу (гемінове залізо), близького за хімічною структурою до простетичних груп гемоглобіну та міоглобіну. За рахунок оберненоїзміни валентності гемінового заліза цитохроми виконують функцію транспорту електронів у ланцюгах біологічного окислення в аеробних клітинах: Залежно від характерних особливостей спектрів поглинання, розрізняють три класи цитохромів (a, b, c). У мітохондріях еукаріотів наявні п’ять різновидів цитохромів — b, c, c1 , a, a3 ; в ендоплазматичному ретикулумі гепатоцитів містяться цитохроми Р-450 та b5 , що беруть участь у реакціях окислювального гідроксилювання. Цитохром (Fe3+) + e– Цитохром (Fe2+)
Молекулярна
організація ланцюга транспорту
електронів (дихального ланцюга)
мітохондрій: послідовність переносників
електронів в дихальному ланцюгу; роль
редокс-потенціалів у транспорті
електронів і протонів.
Локалізація
білків-переносників та коферментів у
дихальному ланцюзі залежить від значення
стандартного окисно-відновного
потенціалу (ОВП) (синонім: редокс-потенціал)
кожного з них. Транспорт електронів
від одного переносника до іншого в
дихальному ланцюзі відбувається в
напрямку зростання ОВП – починається
із субстратів окиснення, які мають
значення редокс-потенціалу в межах
-700 мВ, і закінчується киснем з ОВП +800
мВ.
Чим
більше негативне значення редокс-потенціалу
переносника, тим більша його здатність
віддавати електрони (відновні
властивості). Чим більш позитивне
значення має ОВП переносника, тим більша
його здатність приймати електрони
(окисні властивості).
У 1960-х роках американський вчений Д.
Грін розробив методи виділення та
очищення компонентів дихального ланцюга
та показав, що деякі переносники міцно
зв’язані та виділяються разом – це
комплекси дихального ланцюга. На
сьогодні відомо чотири комплекси (на
рис. 16 вони позначені I,
II,
III,
IV),
які вбудовані в біліпідний шар та
нагадують айсберги, що вільно рухаються
в мембрані й можуть контактувати з
водною фазою матриксу, міжмембранним
простором та між собою (рис. 17). Комплекси
дихального ланцюга: I
комплекс – НАДН-дегідрогеназа
(НАДН-убіхінон оксидоредуктаза) має
дві функції: окислює НАДН.Н+ - переносить
від нього 2 електрони на убіхінон та
транспортує 4Н+ з матриксу у міжмембранний
простір. II
комплекс – сукцинатдегідрогеназа
(сукцинат- убіхінон оксидоредуктаза)
забезпечує додатковий шлях для входу
електронів у дихальний ланцюг за рахунок
окиснення сукцинату. III
комплекс – цитохром с редуктаза
(убіхінон-цитохром с оксидоредуктаза)
містить цитохроми b
та c1,
транспортує 2 електрони з убіхінону на
цитохром с та забезпечує викачування
4 протонів з матриксу в міжмембранний
простір. IV
комплекс – цитохром с оксидаза містить
цитохроми а та а3, транспортує 2 електрони
на кисень та одночасно переносить 2
протони в міжмембранний простір. обота
дихального ланцюга приводить до синтезу
молекул АТФ – це окисне фосфорилювання,
механізми якого будуть розглянуті
нижче. Між тим необхідно звернути увагу
на те, що для синтезу 1 молекули АТФ
потрібно приблизно 32 кДж/моль енергії.
Така енергія виділяється в разі якщо
різниця окисно-відновного потенціалу
між переносниками в дихальному ланцюзі
становить не менше 260 мВ.
Було
встановлено, що в електрон-транспортному
ланцюзі існують три ділянки з таким
перепадом окисно-відновного потенціалу
і вони відповідають комплексам I, II та
IV. Ці ділянки були названі пунктами
спряження з окисним фосфорилюванням,
тобто при транспорті електронів через
ці ділянки виділяється достатньо
енергії для синтезу молекули АТФ (рис.
17).
Електрони,
що транспортуються з НАДН.Н+, проходять
три пункти спряження, тобто виділяється
достатня кількість енергії для синтезу
3 молекул АТФ. Перенесення електронів
з ФАДН2 відбувається лише через два
пункти спряження (рис.
16), тому можливий синтез лише 2 молекул
АТФ. Саме тому завжди реалізація енергії
НАДН.Н+ супроводжується синтезом 3 АТФ,
для ФАДН2 – 2 АТФ.
Надмолекулярні комплекси дихального ланцюга внутрішніх мембран мітохондрій. НАДН-коензим Q-редуктаза — ферментний комплекс (являє собою флаво-протеїн, що містить ФМН), який окислює НАДН і передає відновлювальні еквіваленти на коензим Q(убіхінон); у складі НАДН-коензим Q-редуктази НАДН-дегідрогеназа асоційована з FeS-білками (так званий комплекс І). Сукцинат-коензим Q-редуктаза — ферментний комплекс (ФАД-залежний флавопротеїн), який окислює сукцинат, відновлюючи коензим Q; до складу комплексу входить флавопротеїн сукцинатдегідрогеназа, асоційована з FeS-білком (комплекс II). Коензим Q-цитохром с-редуктаза (убіхінолдегідрогеназа) — ферментний комплекс, що складається з цитохрому b, FeS-білка та цитохрому с1;ферментний комплекс транспортує електрони з відновленого коензиму Q(QH2) на цитохром с (комплекс III). Цитохром с-оксидаза — ферментний комплекс, що складається з цитохромів а та а3 (комплекс IV); комплекс здійснює кінцеву стадію біологічного окислення — відновлення електронами молекулярного кисню; він містить іони міді, як і інші оксид ази.
Шляхи включення відновлювальних еквівалентів (електронів та протонів) у дихальний ланцюг мітохондрій (повний і вкорочений дихальні ланцюги). 1. Включення протонів і електронів у дихальний ланцюг через ФМН флавопротеїну НАДН-коензим Q-редуктази. Цим шляхом на молекулу убіхінону надходять відновлювальні еквіваленти, відщеплені від відповідних субстратів НАДН-залежними дегідрогеназами. 2. Включення протонів і електронів у дихальний ланцюг через ФАД сукцинат-коензим Q-редуктази та деяких інших ФАД-залежних дегідрогеназ. Цим шляхом на убіхінон надходять відновлювальні еквіваленти від янтарної кислоти -метаболіту циклу трикарбонових кислот та певних інших субстратів. Послідовність включення окремих компонентів системи транспорту електронів і протонів у дихальний ланцюг мітохондрій подано на рисунку 1. Як випливає з наведеної схеми, дихальний ланцюг мітохондрій організований таким чином, що перенос у ньому відновлювальних еквівалентів (електронів) відбувається в напрямку від електронегативного кінця (флавопротеїнів) до електропозитивного цитохрому ау Більшість метаболітів (субстрати гліколізу, циклу трикарбонових кислот тощо) передає атоми водню в дихальний ланцюг через НАДН-дегідрогеназу (Е-ФМН); сукцинат та КоА-похідні жирних кислот віддають електрони та протони на коензим Qчерез специфічні флавопротеїни (Е-ФАД), минаючи ФМН-залежний флавопротеїн. Убіхінон є останнім компонентом дихального ланцюга мітохондрій, що здатний транспортувати як електрони, так і протони. На рівні цитохрому bшляхи електронів і протонів розділяються — протони переходять з внутрішньої поверхні мітохондріальної мембрани на зовнішню, а електрони через послідовність цитохромів транспортуються на цитохром ау який відновлює кисень:
![]() Взаємодія
відновленого кисню з вільними протонами
мітохондріального матриксу призводить
до утворення молекули води.
Взаємодія
відновленого кисню з вільними протонами
мітохондріального матриксу призводить
до утворення молекули води.
Шляхи утворення АТФ в клітинах – субстратне та окисне фосфорилування. Утворення АТФ з АДФ та ФН може відбуватися тільки в певних ділянках електронотранспортного ланцюга мітохондрій, в яких величина хімічної енергії, що виділяється при транспортуванні пари електронів між двома редокс-систе- мами (компонентами дихального ланцюга), достатня для синтезу 1 молекули АТФ. Зазначені ділянки електронотранс- портного ланцюга називаютьсяпункта- ми спряження дихання (електронного транспорту) з окисним фосфорилю- ванням

Хеміосмотична теорія окисного фосфорилування – молекулярний механізм генерації АТФ в процесі біологічного окиснення. Хеміоосмотична теорія окисного фосфорилювання Молекулярні механізми генерації АТФ в ході біологічного окислення в мітоходріях пояснюються хеміоосмо- тичною теорією (за П.Мітчелом — P.Mitchell). Головний постулат хеміоосмотичної теорії — спряження електронного транспорту в мітохондріях із біохі- мічною системою синтезу АТФ здійс- нюється за рахунок електрохімічного потенціалу протонів (∆µН+), що утворюється під час функціонування електронотранспортного ланцюга. Хеміоосмотична теорія передба- чає, що: 1. Функціонування дихального (електро- нотранспортного) ланцюга у внутрішніх (спрягаючих) мембранах мітохондрій супроводжується генерацією на цих мембранах електрохімічного градієнта протонів (Н+ ). 2. Окремі компоненти електронотранспортного лан- цюга діють як протонні помпи, що спричиняють век- торний (перпендикулярний площинімембрани) транс- портпротонів, спрямованийунапрямку “матрикс зовнішня поверхня мембрани”. 3. Електрохімічний потенціал протонів на спрягаю- чих мембранах, який створюється завдяки дії протон- них помп дихального ланцюга, є рушійноюсилоюсин- тезу АТФ з АДФ та Фн . 4. Існує ферментна система, що використовує енергію електрохімічного протонного потенціалу для синтезу АТФ за рахунок транслокації протонів через мітохондріальну мембрану в напрямку “зовнішня по- верхня матрикс”. Ця ферментна система, яка замикає протонний цикл на спрягаючих мембранах мітохондрій — про- тонна АТФаза (Н+ –АТФаза), або АТФ-синтетаза. АТФ-синтетаза є білком з четвертинною структурою, що складається з декількох білкових субодиниць, які утворюють компоненти F0 та F1 (F0 F1 – АТФаза). 5. Будь-які фізичні, хімічні та біологічні фактори, що пошкоджують цілісність спрягаючихмембранмітохондрій та розсіюють енергіюелектрохімічного градієнта, порушують синтез АТФ, тобто виступають як роз’єднувачі транс- порту електронів та окисного фос- форилювання. Таким чином, згідно з хеміоос- мотичною теорією, спряження між переносом електронів в дихально- му ланцюзі та синтезом АТФ здійс- нюється за рахунок утворення при функціонуванні протонних помп градієнта концентраціїН+ міждвома поверхнями мітохондріальної мембрани. АТФ-синтетаза, транс- портуючи протони у зворотному напрямку (за електрохімічним гра- дієнтом) призводить до вивіль- нення хімічної енергії, за рахунок якої утворюються макроергічні зв’язки АТФ.
Електрохімічний градієнт протонів (Н+), що утворюється під час функціонування електронно-транспортного ланцюга його роль у спряженні транспорту електронів в мітохондріях з синтезом АТФ. Пункти спряження транспорту електронів та фосфорилування, коефіцієнт окисного фосфоритування АТФ. На думку П. Мітчелла енергія перенесення протонів і електронів у дихальному ланцюзі в першу чергу зосереджується у вигляді протонного потенціалу або електрохімічного градієнта іонів Н+, який створюється перекачуванням протонів через мембрану (по певних пунктах дихального ланцюга) з внутрішньої поверхні внутрішньої мембрани (з боку матриксу) на її зовнішню поверхню - в міжмембранний простір. Одночасно поверхні мембрани заряджаються: зовнішня – позитивно, за рахунок збільшення концентрації Н+, а внутрішня – негативно за рахунок її зменшення і нестачі іонів ОН-, тобто створюється градієнт електричного потенціалу .Отже, ланцюг перенесення електронів працює як протонна помпа, перекачуючи протони з матриксу на зовнішній бік мембрани (рис. 3.10). В результаті цього створюється різниця концентрацій протонів і одночасно різниця електричних потенціалів із знаком плюс на зовнішній поверхні мембрани. Таким чином, на внутрішній мембрані одночасно з градієнтом концентрації протонів і виникає градієнт електричного потенціалу . Розрахунки показали, що дихальний ланцюг мітохондрій при перенесенні двох протонів створює потенціал у 0,25 В. Електрохімічний потенціал, який створився, примушує протони при їх надлишку на зовнішньому боці мембрани рухатися (за градієнтом концентрації) у зворотному напрямку – з зовнішньої мембрани у внутрішню (в матрикс). Проте мембрана є не проникною для них, за винятком окремих ділянок – протонних каналів, які є в ферментативному комплексі Н+-АТФ-синтетази. Зворотна дифузія протонів в матриксі призводить до зрівноваження різниці концентрацій Н+ і відбувається розрядження внутрішньої мембрани (зникає електричний потенціал). Одночасно в активному центрі ферменту Н+_АТФ-синтетази відбувається процес окиснювапьного фосфорилування.
Схема хеміосмотичного механізму спряження транспорту електронів у дихальному ланцюгу з синтезом АТФ. Молекулярна будова та принцип дії АТФ-синтетази. Коефіцієнт окисного фосфорилювання. Регуляція тканинного дихання (дихальний контроль). згідно з хеміоос- мотичною теорією, спряження між переносом електронів в дихально- му ланцюзі та синтезом АТФ здійс- нюється за рахунок утворення при функціонуванні протонних помп градієнта концентраціїН+ міждвома поверхнями мітохондріальної мембрани. АТФ-синтетаза, транс- портуючи протони у зворотному напрямку (за електрохімічним гра- дієнтом) призводить до вивіль- нення хімічної енергії, за рахунок якої утворюються макроергічні зв’язки АТФ. Схему протонного циклу на спрягаючих мембранах мітохонд- рій як рушійної сили окисного фосфорилювання подано на ри- сунку 9.5. Спроможність мітохондрі- альних переносників електронів до транслокації протонів через мембрану зумовлюється особ- ливостями їх внутрішньомемб- ранної топографії. Вважають, що дихальний ланцюг укладений у спрягаючій мембрані у вигляді трьох окислювально-відновлю- вальних “петель”, що відповіда- ють трьом комплексам переносу електронів — I, III та IV. Кожна пара відновлювальних еквіва- лентів (2 Н+ + 2 e– ), проходячи через “петлю”, транспортує два іони Н+ з матриксу в зовнішнє середовище.
Інгібітори транспорту електронів в дихальному ланцюгу мітохондрій. Пояснити механізм дії. Навести приклади. Певні хімічні сполуки здатні специфічним чином порушувати електронний транспорт (інгібітори електронного транспорту) та окисне фосфорилювання (інгібітори та роз’єднувачі окисного фосфорилювання) в мітохондріях. Дані сполуки взаємодіють з певними компонентами дихального ланцюга або систе- мами окисногофосфорилювання, порушуючи їх біохімічніфункції. Інгібітори електронного транспорту Сполуки цього класу порушують функціонування дихального ланцюга міто- хондрій за рахунок зв’язування з окремими ферментними білками або кофермен- тами, що беруть безпосередню участь у переносі електронів від субстратів біо- логічного окислення на О2 . При надходженні в організм людини або тварин ці речовини діють як клітинні отрути, спричиняючи феномен тканинної гіпоксії.
Роз’єднувачі транспорту електронів та окисного фосфорилування в дихальному ланцюгу мітохондрій. Пояснити механізм дії. Навести приклади. . Ротенон — інгібітор транспорту електронів через НАДН-коензим Q-редук- тазний комплекс. Ротенон застосовується як інсектицид. Амобарбітал (амітал) та близький до нього за структурою секобарбітал (секонал). Ці похідні барбітурової кислоти (барбітурати) застосовуються у фарма- кології як снодійні засоби. Разом з тим, барбітурати, подібно до ротенону, є активними інгібіторами клітинного дихання, блокуючи електронний транспорт на рівні НАДН-коензим Q-редуктази. Пієрицидин А — антибіотик, що також блокує НАДН-коензим Q-редуктазний комплекс за рахунок конкурентної взаємодіїз убіхіноном. Антиміцин А — антибіотик, що блокує дихальний ланцюг мітохондрій на рівні переносу електронів через комплекс III (цитохром b — цитохром c1 ). Ціаніди (іони CN – ) — потужні клітинні отрути, що є інгібіторами транспорту електронів на термінальній ділянці дихального ланцюга мітохондрій (у цито- хромоксидазному комплексі). Іони CN – утворюють комплекси з ферри (Fe3+) — формою молекул гему цитохромоксидази, блокуючи їх відновлення до ферро (Fe2+) — форм. Монооксид вуглецю (CO) — інгібірує цитохромоксидазу шляхом зв’язування з ділянкою гему, що взаємодіє з молекулою кисню. Інгібітори окисного фосфорилювання Інгібітори окисного фосфорилювання блокують як окислення субстратів, так і фосфорилювання АДФ у мітохондріях. Олігоміцин — антибіотик, що протидіє як фосфорилюванню АДФ до АТФ, так і стимуляції поглинання О2 , що спостерігається після додавання домітохондрій АДФ (феномен “дихального контролю”). Механізмдії олігоміцину полягає в інгібі- руванні функції АТФ-синтетази.
Активні форми кисню, механізми їх утворення та інактивації. Головним джерелом АФК у клітинах є мітохондрії. Зазвичай приблизно 98%всього кисню, що надходить вклітини, використовується для окислення субстратів зутворенням АТФ і виділенням тепла, і лише 2% використовується в реакціяхосвіти АФК[3], Що може значно зростати при посиленомунадходження кисню в клітини або порушенні роботи електронно-транспортного ланцюга мітохондрій. Ю.А. Владимиров[4]виділяє три категорії АФК: первинні, вторинні ітретинні. Первинні АФК утворюються при окисленні деяких молекул і володіютьрегуляторним абопомірним антимікробну дію. До них відносяться оксид азотуNO, що володіє судинорозширювальну дію, і супероксид ГО -, Доляякого може бути досить різноманітною. Зазвичай за допомогою спеціалізованогоферменту супероксиддисмутази він перетворюється на перекис водню Н 2. Про 2. і надалі - в гіпохлорит ClO -. Обидва ці сполуки використовуютьсямакрофагами для боротьби з бактеріями. Принедостатньою нейтралізації супероксидуйого надлишок, взаємодіючи з NO, утворює пероксинітрит або переводитьтривалентне залізо Fe 3 +. в двовалентне Fe 2 +., Якепри взаємодії з Н 2. Про 2., НClО і ліпоперекісямі утворюєгідроксильний радикал ОН * або ліпоксільний радикал LO *.Ці радикали, як і пероксинітрит, представляють категорію вториннихрадикалів,саме ця категорія має сильний токсичною дією внаслідок своєїздібності необоротно пошкоджувати мембранні ліпіди, а також молекули ДНК,вуглеводів і білків.
Намалювати та пояснити графіки залежності активності пепсину та амілази слини від рН середовища із зазначенням рН оптимуму цих ферментів. Якщо активність ферментів визначати при різних значення рН, то графік залежності матиме вигляд куполоподібної кривої (рис. ), крива може бути симетрична, з однаковим підйомом і спуском у кислому і лужному середовищах, або йти на зниження в якійсь одній ділянці. Значення рН, при якому активність ферменту найвища, називається рН-оптимумом ферменту. Звичайно ферменти найактивніші в межах вузької зони рН, яка для більшості з них знаходиться в межах рН 6‑8. Ряд внутрішньоклітинних ферментів найкраще функціонує у нейтральному середовищі. Разом із тим, для ферментів шлунково-кишкового тракту рН-оптимум може знаходитись у зоні від нейтрального до сильно кислого і лужного середовищ. Так, пепсин має оптимум рН 1,5‑2,5, трипсин – рН 8,0-9,0. Для виявлення залежності активності ферменту від концентрації водневих іонів експерименти проводять при оптимальній температурі, достатньо високих концентраціях субстрату, але при різних значеннях рН. Чутливість активності ферментів до зміни рН середовища, очевидно, пов’язана з тим, що фермент, залежно від середовища, може знаходитись в іонізованій або неіонізованій формі, що буде обов’язково відображатися на третинній структурі білка, зокрема при формуванні активного центру та утворенні фермент-субстратного комплексу. Безумовно, різне значення рН буде по-різному впливати і на стан іонізації кофакторів та субстратів. Можна стверджувати, що саме при рН-оптимумі між субстратом і ферментом існує найкраща просторова та електростатична комплементарність, яка забезпечує їх зв’язування, утворення фермент-субстратного комплексу і подальше перетворення його. На рис. показана графічна залежність активності ферментів від рН середовища.

Дослідити процес окисного фосфорилування в мітохондріях, пояснити, на чому він базується. Які фармакологічні та фізіологічні сполуки є роз’єднувачами дихання і фосфорилування? Пояснити біохімічний механізм їх дії. Певні хімічні сполуки здатні специфічним чином порушувати електронний транспорт (інгібітори електронного транспорту) та окисне фосфорилювання (інгібітори та роз’єднувачі окисного фосфорилювання) в мітохондріях. Дані сполуки взаємодіють з певними компонентами дихального ланцюга або систе- мами окисногофосфорилювання, порушуючи їх біохімічніфункції. Інгібітори електронного транспорту Сполуки цього класу порушують функціонування дихального ланцюга міто- хондрій за рахунок зв’язування з окремими ферментними білками або кофермен- тами, що беруть безпосередню участь у переносі електронів від субстратів біо- логічного окислення на О2 . При надходженні в організм людини або тварин ці речовини діють як клітинні отрути, спричиняючи феномен тканинної гіпоксії. Ротенон — інгібітор транспорту електронів через НАДН-коензим Q-редук- тазний комплекс. Ротенон застосовується як інсектицид. Амобарбітал (амітал) та близький до нього за структурою секобарбітал (секонал). Ці похідні барбітурової кислоти (барбітурати) застосовуються у фарма- кології як снодійні засоби. Разом з тим, барбітурати, подібно до ротенону, є активними інгібіторами клітинного дихання, блокуючи електронний транспорт на рівні НАДН-коензим Q-редуктази. Пієрицидин А — антибіотик, що також блокує НАДН-коензим Q-редуктазний комплекс за рахунок конкурентної взаємодіїз убіхіноном. Антиміцин А — антибіотик, що блокує дихальний ланцюг мітохондрій на рівні переносу електронів через комплекс III (цитохром b — цитохром c1 ). Ціаніди (іони CN – ) — потужні клітинні отрути, що є інгібіторами транспорту електронів на термінальній ділянці дихального ланцюга мітохондрій (у цито- хромоксидазному комплексі). Іони CN – утворюють комплекси з ферри (Fe3+) — формою молекул гему цитохромоксидази, блокуючи їх відновлення до ферро (Fe2+) — форм. Монооксид вуглецю (CO) — інгібірує цитохромоксидазу шляхом зв’язування з ділянкою гему, що взаємодіє з молекулою кисню. Інгібітори окисного фосфорилювання Інгібітори окисного фосфорилювання блокують як окислення субстратів, так і фосфорилювання АДФ у мітохондріях. Олігоміцин — антибіотик, що протидіє як фосфорилюванню АДФ до АТФ, так і стимуляції поглинання О2 , що спостерігається після додавання домітохондрій АДФ (феномен “дихального контролю”). Механізмдії олігоміцину полягає в інгібі- руванні функції АТФ-синтетази. Роз’єднувачі окисного фосфорилювання Сполуки цього класу спричиняють “неконтрольоване” дихання мітохондрій, яке не залежить від функціонування системи фосфорилювання АДФ. В присут- ності роз’єднувачів спостерігається активне поглинання мітохондріями О2 , незва- жаючи на зниження швидкості (або відсутність) генерації АТФ з АДФ та Фн . Згідно з хеміоосмотичною теорією, роз’єднувачі спричиняють втрату мембраною протонного потенціалу — рушійної сили генерації макроергічних зв’язків АТФ. До роз’єднувачів окисного фосфорилювання належать: – 2,4-динітрофенол та сполуки, близькі до нього за хімічною структурою (динітрокрезол, пентахлорфенол); – СССР (карбонілціанід-м-хлорфенілгідразон) — сполука, що в 100 разів пере- вищує за специфічною активністю 2,4-динітрофенол. Здатність роз’єднувати дихання та окисне фосфорилювання в мітохондріях мають такожгормони щитовидноїзалози (тироксин, трийодтиронін).
Анаеробне окиснення глюкози – гліколіз: ферментативні реакції гліколізу, енергетика, регуляція. Гліколітична оксидоредукція, субстратне фосфорилування в гліколізі. За умов анаеробного гліколізу (наприклад, в інтенсивно працюючих скелетних м'язах або в молочнокислих бактеріях) гліколі-тичний НАДН не віддає свої відновлювальні еквіваленти в дихальний ланцюг мітохондрій, а використовується для відновлення пірувату до L-лактату: Реакція каталізується ферментом лактатдегідрогеназою, яка існує у вигляді п яти різних ізоферментних форм (ЛДГ1—ЛДГ5), що відрізняються за своїми кінетичними властивостями (КМ, Vмах, ступенем алостеричного інгібірування піруватом). Таким чином, ферментативні реакції анаеробного гліколізу майже повністю спів¬падають із реакціями аеробного гліколізу, відрізняючись лише на етапі, що відбувається після утворення пірувату: при аеробному гліколізі піруват є субстратом перетворення на ацетилкоензим А та подальшого окислення, а при анаеробному гліколізі піруват відновлюється до лактату за рахунок НАДН, що утворився в реакціях гліколітичної оксидоредукції. Інакше кажучи, після утво¬рення пірувату подальше його перетворення може відбуватися за одним із двох альтер¬нативних шляхів, що залежать від стану окислювально-відновлювальних процесів у певній тканині: - в аеробних умовах відбувається окисне декарбоксилювання пірувату доацетил-КоА, який у подальшому окислюється до СО, та Н,О в циклі Кребса; НАДН, що утворився при окисленні гліцеральдегід-3-фосфату, віддає свої відновлювальні еквіваленти на дихальний ланцюг мітохондрій через спеці¬альні човникові механізми; - в анаеробнихумовах (або в умовах гіпоксії) реокислення гліколітичного НАДН відбува¬ється за рахунок дії лактатдегідрогенази, яка відновлює піруват до лактату; течія лактат-дегідрогеназної реакції в даному напрямку генерує НАД+, що знову використовується для окислення гліцеральдегід-3-фосфату і подальшого накопичення лактату як про¬дукту анаеробного гліколізу. Така послідовністьреакцій найбільш характерна для інтенсивно працюючихскелетних м'язів; крім скелетних м'язів та еритроцитів, клітини деяких інших органів та тканин (головного мозку, шлунково-кишкового тракту, мозкового шару нирок, сітківки та шкіри) частково задовольняють свої енергетичні потреби за рахунок анаероб¬ного гліколізу, утворюючи молочну кислоту.

Вкдад робіт Ембдена, Мейєргофа, Парнаса у встановлення послідовності ферментативних реакцій гліколізу (молочнокислого бродіння). Молочнокисле бродіння — процес анаеробного окислення вуглеводів, кінцевим продуктом при якому виступає молочна кислота. Назва отримана по характерному продукту — молочної кислоти. Для молочнокислих бактерій є основним шляхом катаболізму вуглеводів і основним джерелом енергії у вигляді АТФ. Також молочнокисле бродіння відбувається в тканинах тварин у відсутності кисню при великих навантаженнях. Розрізняють такі назви гомоферментативне і гетероферментативне молочнокисле бродіння, в залежності від продуктів що виділяються, крім молочної кислоти та їх відсоткового співвідношення. Відмінність також полягає й у різних шляхах одержання пірувату при деградації вуглеводів гомо-і гетероферментативними молочнокислими бактеріями. Гомоферментативне молочнокисле бродіння. При гомоферментативне молочнокислому бродінні вуглевод спочатку окислюється до пірувату по гліколітичному шляху, потім піруват відновлюється до молочної кислоти НАДН + Н (утворився на стадії гліколізу при дегідруванні гліцеральдегід-3-фосфата) за допомогою лактатдегідрогенази. Від стереоспеціфічності лактатдегідрогенази та наявності лактатрацемази залежить, який енантіомер молочної кислоти буде превалювати в продуктах: L-, D-молочна кислота або ж DL-рацемат. Продуктом гомоферментативного молочнокислого бродіння є молочна кислота, яка становить не менше 90% всіх продуктів бродіння. Приклади гомоферментативних молочнокислих бактерій:Lactobacillus casei, L. acidophilus, Streptococcus lactis . Гетероферментативне молочнокисле бродіння. На відміну від гомоферментативного бродіння, деградація глюкози йде по пентозофосфатному шляху, гліцеральдегид-3-фосфат що утворюється з ксилулозо-5-фосфату окислюється до молочної кислоти, а ацетілфосфат відновлюється до етанолу (деякі гетероферментативні молочнокислі бактерії окислюють отриманий етанол частково або повністю до ацетату). Таким чином, при гетероферментативному молочнокислому бродінні утворюється більше продуктів: молочна кислота, оцтова кислота, етанол, двоокис вуглецю. приклади гетероферментативних молочнокислих бактерій: L. fermentum , L. brevis , Leuconostoc mesenteroides , Oenococcus oeni. Значення молочнокислого бродіння для людини. Молочнокисле бродіння використовується для консервації продуктів харчування (за рахунок інгібування росту мікроорганізмів молочною кислотою і зниження рН) з метою тривалого збереження (приклад — квашення овочів, сирокопчення), приготуванні кисломолочних продуктів (кефіру , ряжанки , йогурту, сметани), силосуванні рослинної маси, а також біотехнологічного способу виробництва молочної кислоти.
Спиртове бродіння, ферментативні реакції. Спиртове бродіння — біохімічний процес ферментації, при якому цукри, такі як глюкоза і фруктоза, розкладаються під дією ферментів з виділенням енергії і утворенням етилового спирту та вуглекислого газу. Дозволяє отримати два моль АТФ на моль глюкози в анаеробних умовах. Загальне рівняння спиртового бродіння: C6H12O6 + 2 АДФ + 2 Фн → 2 C2H5OH + 2 CO2 + 2 АТФ + 2 H2O Цей метаболічний шлях характерний для багатьох грибів водоростей, найпростіших та деяких бактерій. Спиртове бродіння здавна використовується людиною у процесі хлібопекарства (спричиняє «сходження» дріжджового тіста) та виготовлення алкогольних напоїв. Під час спиртового бродіння розщеплення глюкози починається гліколітичним шляхом (за винятком бактерії Zymomonas mobilis, у якої глюкоза метаболізує по шляху Ентнера-Дудорова[1]). У гліколітичних реакціях глюкоза розщеплюється і окиснюється до двох молекул пірувату, відбувається субстратне фосфорилювання двох молекул АДФ із утворенням АТФ, а також відновлюються до НАДH дві молекули НАД+. За аеробних умов НАДH знову окиснюється віддаючи електрони через ряд посередників на молекулярний кисень, і тоді знову може бути використаний у процесі гліколізу. В анаеробних умовах регенерація НАД+ відбувається у кінцевих етапах бродіння, під час яких акцептором електронів є сам піруват або його похідні: у випадку спиртового бродіння — ацетальдегід. Ацетальдегід утворюється з пірувату шляхом декарбоксилювання (відщеплення вуглекислого газу), яке каталізується піруватдекарбоксилазою. Цей фермент потребує присутності іонів Mg2+ та містить ковалентно приєднаний кофермент тіамінпірофосфат[3]. Наступним кроком є відновлення ацетальдегіду до етилового спирту завдяки перенесенню гідрид іона із НАДH, утвореного у гліколізі[4]. Реакція відбувається за участі ферменту алкогольдегідрогенази, що містить в активному центрі іон цинку, який поляризує карбонільну групу субстрату полегшуючи приєднання гідриду[3][5]. Отже кінцевими продуктами спиртового бродіння на одну молекулу глюкози є дві молекули етилового спирту, дві молекули CO2, та дві молекули АТФ. В підсумку не відбувається ні окиснення ні відновлення глюкози (співвідношення C:H однакове для вихідних речовин (глюкоза) і продуктів (етанол + вуглекислий газ) і становить 1:2)[6][5].

Етапи
аеробного окиснення глюкози 1.
Розщеплення глюкози до піровиноградної
кислоти. Продуктом цього гліколітичного
етапу розщеплення глюкози є піруват,
а також дві молекули відновленого НАД+
і дві молекули АТФ:
![]() 2.
Окислювальне декарбоксилювання
піровиноградної кислоти. У результаті
цього процесу утворюється ацетилкоензим
А — основний субстрат окислення в циклі
трикарбонових кислот — та відновлена
форма НАД+ . Сумарне рівняння окислювального
декарбоксилювання пірувату:
2.
Окислювальне декарбоксилювання
піровиноградної кислоти. У результаті
цього процесу утворюється ацетилкоензим
А — основний субстрат окислення в циклі
трикарбонових кислот — та відновлена
форма НАД+ . Сумарне рівняння окислювального
декарбоксилювання пірувату:
![]() Окислювальнедекарбоксилюванняпірувату
каталізуєтьсяпіруватдегідрогеназним
комплексом — мультиферментною системою,
яка в клітинах еукаріотів міститься в
мембранах мітохондрій, а у прокаріотів
— у цитоплазмі. До складу цього комплексу
входять три ферменти, що каталізують
три послідовні стадії перетворення
пірувату на ацетил-КоА: піруватдегідрогеназа,
дигідроліпоїлацетилтрансфераза,
дигідро- ліпоїлдегідрогеназа та п’ять
коферментів і простетичних груп:
тіаміндифосфат (ТДФ), коензим А (КоА),
ліпоєва кислота (ЛК), НАД+ , ФАД.
Окислювальнедекарбоксилюванняпірувату
каталізуєтьсяпіруватдегідрогеназним
комплексом — мультиферментною системою,
яка в клітинах еукаріотів міститься в
мембранах мітохондрій, а у прокаріотів
— у цитоплазмі. До складу цього комплексу
входять три ферменти, що каталізують
три послідовні стадії перетворення
пірувату на ацетил-КоА: піруватдегідрогеназа,
дигідроліпоїлацетилтрансфераза,
дигідро- ліпоїлдегідрогеназа та п’ять
коферментів і простетичних груп:
тіаміндифосфат (ТДФ), коензим А (КоА),
ліпоєва кислота (ЛК), НАД+ , ФАД.
Окиснювальне декарбоксилування пірувату, мультиферментний піруватдегідрогеназний комплекс – особливості функціонування за участю трьох ферментів та п’яти коферментів. Процес окислювального декарбоксилування пірувату включає реакції дегідрування та декарбоксилування, у ході яких карбоксильна група вивільняється у вигляді СО2, а ацетильний залишок переноситься на коензим А: Піруват + НАД+ + НS-КоА Ацетил-КоА + СО2 + НАДН + Н+ Сукупність цих реакцій каталізує піруватдегідрогеназний комплекс. він міститься в мітохондріях, тому піруват, який утворюється в процесі гліколізу в цитоплазмі, мусить переміститися туди. Піруватдегідрогеназний комплекс являє собою мультиферментну систему з М.м. 4·106 Да і складається з трьох ферментів: піруватдегідрогенази, дигідроліпоїлдегідрогенази та дигідроліпоїлацетилтрансферази (рис. 4.7). Ці ферменти двокомпонентні та містять 5 коферментів: НАД+, ФАД, тіаміндифосфат (ТДФ) або тіамінпірофосфат (ТПФ), ліпоєву кислоту і кофермент ацилювання (КоА-SН). При окисненні пірувату утворюється не вільна ацетатна кислота, а так звана “активна” або “активована” ацетатна кислота у вигляді ацетил-КоА. Остання за рахунок наявності макроергічного зв'язку може бути використана в різних синтетичних процесах. Докладне дослідження окиснення пірувату за участю дегідрогенази показало, що реакція відбувається в декілька етапів (для спрощення органічні залишки в молекулі ТДФ позначимо радикалами R1, R2, R3): Перший етап – це взаємодія пірувату та ТДФ з утворенням проміжної сполуки – активного пірувату, який, декарбоксилуючись, під дією ферменту піруватдекарбоксилази перетворюється на оксіетилтіаміндифосфатОксіетил-ТДФ реагує з окисненою формою ліпоєвої кислоти, яка входить до складу ліпоаттрансацетилази, при цьому утворюється ацетил-ТДФ На другому етапі за участю ферменту дигідроліпоїлацетилтрансферази залишок ацетату переноситься з ацетил–ТДФ на другу молекулу ліпоєвої кислоти з утворенням ацетил–гідроліпоату: дигідроліпоїлацетилтрансфераза каталізує і третю стадію – перенесення ацетильної групи на КоА-SH з утворенням кінцевого продукту ацетил-КоА, який є високоенергетичною (макроергічною) сполукою. На четвертому етапі відбувається зворотне окиснення (реоксидація) аміду дигідроліпоєвої кислоти відбувається за участю ферменту дигідроліпоїлдегідрогенази, яка коферментом містить ФАД. Амід дигідроліпоєвої кислоти окиснюється до аміду ліпоєвої кислоти. Атоми водню передаються на ФАД через третю молекулу ліпоєвої кислоти, ФАД при цьому відновлюється. Від ФАДН2 за участі дигідроліпоїлдегідрогенази атоми водню відразу ж переходять на окиснену форму НАД+, тому в сумарному рівнянні реакції окиснення пірувату в якості акцептора водню виступає НАД+ НАДН окиснюється киснем за схемою: Ацетил-КоА, конденсуючись із оксалоацетатом, перетворюється на лимонну кислоту, яка є одним з компонентів циклу ди- і трикарбонових кислот (циклу лимонної кислоти). За допомогою цього циклу та цитохромної системи ацетил-КоА в результаті низки перетворень окиснюється до кінцевих продуктів СО2 і Н2О. Вивільнений при цьому КоА-SH може вступати в реакцію з наступними молекулами аміду ацетилліпоєвої кислоти.
Порівняльна характеристика біоенергетики аеробного та анаеробного окиснення глюкози. У процесі анаеробного окислення глюкози витрачається 2 молекули АТФ (при фосфорилюванні глюкози з утворенням глюкозо-6-фосфату і при перетворенні фруктозо-6-фосфату на фруктозо-1,6-дифосфат), а синтезується 4 АТФ (по дві у реакціях гліцеральдегід-3-фосфат → 3-фосфогліцерат та фосфоенолпіруват → піруват). Різниця у затраченій і утвореній кількості АТФ складає +2 молекули. Сумарне рівняння: С6Н12О6 + 2АДФ + 2Фн→ 2С3Н4О3(С3Н6О3) + 2АТФ. При аеробному окисленні глюкози (повне окислення до СО2 і Н2О): генерується 2АТФ на етапі аеробного гліколізу; гліколітичний НАДН за рахунок окислення в мітохондріях дає 2НАДН*3АТФ= 6АТФ; окисне декарбоксилювання ПВК дає 2НАДН, які в мітохондріях окислються з утворенням 2НАДН*3АТФ= 6АТФ; в ЦТК 2 ацетил-КоА (із попереднього декарбоксилування ПВК) дають 2ац-КоА*12АТФ=24АТФ. Сумарне рівняння: С6Н12О6 + 6О2 + 38АДФ + 38Фн→ 6СО2 + 6Н2О + 38АТФ. Ефект Пастера – не утворення лактату в присутності О2, через те, що в умовах активного клітинного дихання пригнічується активність фосфофруктокінази і піруваткінази.
Човникові
механізми переносу гліколітичного
НАДН. Малат-аспартатний та гліцерофосфатний
шунти транспорту відновлювальних
еквівалентів гліколітичного НАДН в
мітохондрії в аеробних умовах. У
процесі перетворення гліцеральдегід-3–фосфату
утворюється дві молекули НАДН, які при
окисненні дають не 6 молекул АТФ, а лише
4. Це відбувається тому, що молекули
позамітохондріального НАДН не здатні
проникати всередину мітохондрій, але
електрони, які вони віддають, можуть
включатися у мітохондріальний ланцюг
біологічного окиснення за допомогою
гліцерофосфатного
човникового
механізму. Як видно з рис. 4.5, цитоплазматичний
НАДН спочатку реагує з цитоплазматичним
діоксіацетонфосфатом, утворюючи
гліцерол-3-фосфат. Реакція каталізується
НАД-залежною цитоплазматичною
гліцерол-3-фосфат-дегідрогеназою Утворений
гліцерол-3-фосфат легко проникає через
мітохондріальну мембрану. Всередині
мітохондрії інша (мітохондріальна)
гліцерол-3-фосфат-дегідрогеназа
знову окиснює гліцерол-3–фосфат до
діоксіацетонфосфату:
Утворений
гліцерол-3-фосфат легко проникає через
мітохондріальну мембрану. Всередині
мітохондрії інша (мітохондріальна)
гліцерол-3-фосфат-дегідрогеназа
знову окиснює гліцерол-3–фосфат до
діоксіацетонфосфату:
 Відновлений
флавопротеїн передає за допомогою КоQ
отримані ним електрони в ланцюг
біологічного окиснення та спряженого
з ним окисного фосфорилування, а
діоксіацетонфосфат виходить із
мітохондрій у цитоплазму і може знову
взаємодіяти з цитоплазматичним НАДН.
Таким чином, пара електронів (із 1
молекули цитоплазматичного НАДН), що
передається у дихальний ланцюг за
допомогою гліцерофосфатного човникового
механізму дає не 3 АТФ, а 2 молекули АТФ.
Така човникова система перенесення
відновлених еквівалентів від цитозольного
НАДН
у
мітохондрії існує тільки у скелетних
м'язах і мозку. У клітинах печінки, нирок
і серця діє більш складний малат-аспартатний
човниковий механізм, який забезпечує
утворення 3 молекул АТФ на одну молекулу
цитоплазматичного НАДН (рис. 4.6). Дія
такого човникового механізму стає
можливою завдяки присутності
малатдегідрогенази
та аспартатамінотрансферази
як в цитозолі, так і в мітохондріях.
Встановлено, що спочатку від цитозольного
НАДН відновлювальні еквіваленти за
участю малатдегідрогенази
переносяться на цитозольний оксалоацетат.
У результаті утворюється малат. останній
за допомогою системи, що транспортує
дикарбонові кислоти, проникає через
внутрішню мембрану мітохондрій у
матрикс. Тут малат окиснюється до
оксалоацетату, а матриксний НАД+
відновлюється до НАДН, який може тепер
передавати свої електрони на ланцюг
дихальних ферментів, що локалізований
на внутрішній мембрані мітохондрій. У
свою чергу, утворений оксалоацетат у
присутності глутамату і фермента АсАТ
вступає в реакцію трансамінування.
Утворені аспарат і α-кетоглутарат за
допомогою спеціальних транспортних
систем здатні переноситися через
мембрану мітохондрій. Трансамінування
в цитозолі регенерує оксалоацетат,
запускає наступний цикл. У цілому процес
включає зворотні реакції, відбувається
без затрати енергії,"рушійною силою"
його є постійне відновлення НАД+
у цитозолі гліцеральдегід-3-фосфатом,
який утворюється при катаболізмі
глюкози. Завдяки функціонуванню
малат-аспартатного човникового механізму
в результаті повного окиснення 1 молекули
глюкози може утворитися не 36, а 38 молекул
АТФ.
Відновлений
флавопротеїн передає за допомогою КоQ
отримані ним електрони в ланцюг
біологічного окиснення та спряженого
з ним окисного фосфорилування, а
діоксіацетонфосфат виходить із
мітохондрій у цитоплазму і може знову
взаємодіяти з цитоплазматичним НАДН.
Таким чином, пара електронів (із 1
молекули цитоплазматичного НАДН), що
передається у дихальний ланцюг за
допомогою гліцерофосфатного човникового
механізму дає не 3 АТФ, а 2 молекули АТФ.
Така човникова система перенесення
відновлених еквівалентів від цитозольного
НАДН
у
мітохондрії існує тільки у скелетних
м'язах і мозку. У клітинах печінки, нирок
і серця діє більш складний малат-аспартатний
човниковий механізм, який забезпечує
утворення 3 молекул АТФ на одну молекулу
цитоплазматичного НАДН (рис. 4.6). Дія
такого човникового механізму стає
можливою завдяки присутності
малатдегідрогенази
та аспартатамінотрансферази
як в цитозолі, так і в мітохондріях.
Встановлено, що спочатку від цитозольного
НАДН відновлювальні еквіваленти за
участю малатдегідрогенази
переносяться на цитозольний оксалоацетат.
У результаті утворюється малат. останній
за допомогою системи, що транспортує
дикарбонові кислоти, проникає через
внутрішню мембрану мітохондрій у
матрикс. Тут малат окиснюється до
оксалоацетату, а матриксний НАД+
відновлюється до НАДН, який може тепер
передавати свої електрони на ланцюг
дихальних ферментів, що локалізований
на внутрішній мембрані мітохондрій. У
свою чергу, утворений оксалоацетат у
присутності глутамату і фермента АсАТ
вступає в реакцію трансамінування.
Утворені аспарат і α-кетоглутарат за
допомогою спеціальних транспортних
систем здатні переноситися через
мембрану мітохондрій. Трансамінування
в цитозолі регенерує оксалоацетат,
запускає наступний цикл. У цілому процес
включає зворотні реакції, відбувається
без затрати енергії,"рушійною силою"
його є постійне відновлення НАД+
у цитозолі гліцеральдегід-3-фосфатом,
який утворюється при катаболізмі
глюкози. Завдяки функціонуванню
малат-аспартатного човникового механізму
в результаті повного окиснення 1 молекули
глюкози може утворитися не 36, а 38 молекул
АТФ.
Пентозофосфатний шлях (ПФШ) окиснення глюкози; схема, біологічне значення, особливості функціонування в різних тканинах. Послідовність ферментативних реакцій ПФШ, окислювальна стадія та стадія ізомерних перетворень пентозо-, гексозо- та гептозофосфатів. Значення ПФШ як донора НАДФН у відновлювальному синтезі жирних кислот та стероїдів, як постачальника рибозо-5-фосфату для утворення нуклеотидів у синтезі нуклеїнових кислот. Можливість існування прямого окиснення глюкози без попереднього розщеплення на фосфотріози була встановлена дослідженнями О. Варбурга та В.О. Енгельгардта. Основний шлях катаболізму глюкози в організмі людини і тварин — це поєднання гліколізу та циклу лимонної кислоти. Проте існують альтернативні шляхи, які виконують специфічні функції. Один із них (пентозофосфатний) включає перетворення глюкозо-6-фосфату на пентозофосфати і СО2 і в такий спосіб забезпечує клітини рибозо-5-фосфатом для синтезу нуклеотидів і нуклеїнових кислот. Крім цього, ПФШ постачає відновлену форму НАДФН, необхідну для реакцій відновлення під час синтезу жирних кислот і стероїдних сполук, для мікросомального окиснення. Усі реакції відбуваються в цитоплазмі клітин. ПФШ ще називають пентозофосфатним циклом (ПФЦ). У ПФШ умовно виділяють дві стадії – окисну та неокисну (рис. 4.19). Окисна стадія розпочинається окисненням глюкозо-6-фосфату з утворенням НАДФН і 6-фосфоглюконолактону, який гідролізується до 6-фосфоглюконової кислоти (тому ПФШ називають також фосфоглюконатним) з подальшим її дегідруванням і декарбоксилуванням з утворенням пентози (рибулозо-5-фосфату). Ці реакції окисної стадії незворотні. Неокисна стадія ПФШ включає зворотні реакції ізомеризації пентоз через проміжні сполуки — гептозу (седогептулозо-7-фосфат), тетрозу (еритрозо-4-фосфат), тріозу (гліцеральдегід-3-фосфат) і перетворення їх на фруктозо-6-фосфат. Утворений фруктозо-6-фосфат перетворюється на глюкозо-6-фосфат або розпадається шляхом гліколізу. Таким чином ПФШ може переходити в гліколітичний. Так здійснюється утилізація пентоз, утворених в окисній стадії ПФШ, і рибозо-5-фосфату, утвореного при розпаді нуклеїнових кислот. Реакції неокисної стадії ПФШ перебігають і у зворотному напрямку. За рахунок цього пентози можуть синтезуватись із глюкози не через окисну стадію ПФШ, а із проміжних продуктів гліколізу – фруктозо-6-фосфату і гліцеральдегід-3-фосфату. Сумарний перехід п'яти молекул фруктозо-6-фосфату дасть шість молекул рибозо-5-фосфату. Такий напрямок реакцій неокисної стадії ПФШ має місце в тканинах, в яких потреба в пентозах більша, ніж потреба в НАДФН.







Порушення пентозофосфатного шляху в еритроцитах: ензимопатії глюкозо-6-фосфат-дегідрогенази. Відомо приблизно 80 варіантів глюкозо-6-фосфатдегідрогенази еритроцитів, які відрізняються за каталітичною активністю, спорідненістю до субстратів, чутливістю до температури, рН, інгібіторів, електрофоретичною рухливістю. Активність ферменту може бути зниженою різною мірою, аж до нульової. Наявність у людини варіанту глюкозо-6-фосфатдегідрогенази з активністю, близькою до нуля, клінічно проявляється гемолітичною анемією. Механізм розвитку гемолітичної анемії полягає в наступному: в еритроцитах НАДФ+ переводить окиснений глутатіон у відновлений. Останній попереджує пероксидне окиснення ліпідів мембран, захищає від окиснення SН-групи білків, підтримує відновлений стан заліза в гемоглобіні. Знижена продукція НАДФН внаслідок вираженої недостатності глюкозо-6-фосфатдегідрогенази зумовлює зниження рівня в еритроцитах відновленого глутатіону, накопичення Н2О2 і, як наслідок, гемоліз еритроцитів. При вираженій недостатності ферменту (активність нижча 10 %) анемія розвивається під впливом деяких лікарських засобів, які володіють властивостями оксидантів (протималярійних, сульфаніламідних тощо). Або після споживання в їжу бобів Vicia Faba (кінські боби). У людей із менш вираженою недостатністю ферменту клінічні прояви, як правило, відсутні.
Метаболічний
шлях та ферментативні реакції перетворення
фруктози в організмі людини. Спадкові
ензимопатії пов’язані з генетичними
дефектами синтезу ферментів метаболізму
фруктози – несприйнятність фруктози,
фруктоземія. Важливими
субстратами енергетичного обміну є
моносахариди – фруктоза та галактоза,
які надходять в організм людини переважно
з харчовими дисахаридами рослинного
і тваринного походження: фруктоза – у
складі сахарози, що гідролізується в
кишечнику ферментом сахаразою до
глюкози і фруктози, галактоза – в складі
“молочного цукру” лактози, що
розщеплюється кишковою лактазою до
галактози та глюкози. Після проникнення
через плазматичні мембрани всередину
клітин фруктоза та галактоза включаються
в обмінні процеси шляхом їх перетворення
на фосфорильовані ефіри глюкози або
її метаболітів – інтермедіатів
гліколізу.
Основним
джерелом фруктози в організмі людини
є її надходження з продуктами харчування,
певна кількість синтезується в
спеціалізованих тканинах ( у сім'яних
везикулах). Її концентрація в сім'яній
рідині досягає 10 ммоль. За рахунок
здатності мітохондрій сперматозоїдів
окиснювати фруктозу в процесі фруктолізу,
вона є енергетичним джерелом для руху
сперматозоїдів.
У
певній концентрації фруктоза міститься
в кришталику ока, де вона також утворюється
з глюкози через сорбітол. Активність
цього метаболічного шляху збільшується
при цукровому діабеті; оскільки
проникнення сорбітолу через клітинні
мембрани є утрудненим, він разом із
фруктозою нагромаджується в кришталику,
що становить біохімічну основу розвитку
діабетичної катаракти.
Включення
фруктози в обмін глюкози здійснюється
двома шляхами:
1.
Під впливом неспецифічної гексокінази,
що каталізує фосфорилування різних
гексоз: Утворений
D-фруктозо-6-фосфат є метаболітом
гліколітичного окиснення глюкози. Цей
шлях метаболізму фруктози реалізується
в клітинах багатьох
органів, особливо в м'язах і нирках. 2.
Під впливом специфічної фруктокінази,
яка каталізує фосфорилування фруктози
не за 6-м, а за 1-м вуглецевим атомом (рис.
4.20):
Утворений
D-фруктозо-6-фосфат є метаболітом
гліколітичного окиснення глюкози. Цей
шлях метаболізму фруктози реалізується
в клітинах багатьох
органів, особливо в м'язах і нирках. 2.
Під впливом специфічної фруктокінази,
яка каталізує фосфорилування фруктози
не за 6-м, а за 1-м вуглецевим атомом (рис.
4.20):
 Цей
шлях перетворення фруктози функціонує
в печінці.
Утворений
D-фруктозо-1-фосфат розщеплюється під
дією альдолази (фруктозо-1-фосфатальдолази)
на дві тріози:
Цей
шлях перетворення фруктози функціонує
в печінці.
Утворений
D-фруктозо-1-фосфат розщеплюється під
дією альдолази (фруктозо-1-фосфатальдолази)
на дві тріози:
 Діоксіацетонфосфат,
що утворився, є інтермедіатом гліколізу
і перетворюється на гліцеральдегід-3-фосфат
при дії тріозофосфатізомерази Інший
продукт альдолазної реакції,
гліцеральдегід, фосфорилується також
з утворенням гліцеральдегід-3-фосфату
(фермент — тріозокіназа)
Діоксіацетонфосфат,
що утворився, є інтермедіатом гліколізу
і перетворюється на гліцеральдегід-3-фосфат
при дії тріозофосфатізомерази Інший
продукт альдолазної реакції,
гліцеральдегід, фосфорилується також
з утворенням гліцеральдегід-3-фосфату
(фермент — тріозокіназа) Таким чином, фруктокіназний шлях обміну
фруктози перетинається з гліколізом
на етапі утворення двох молекул
фосфотріози — гліцеральдегід-3-фосфату.
Оскільки перетворення фруктози на
тріози здійснюється без участі двох
регуляторних реакцій гліколізу
(гексокіназної та фосфофруктокіназної),
які обмежують швидкість процесу,
катаболізм фруктози відбувається
значно швидше порівняно з глюкозою.
Синтез ендогенної фруктози відбувається
через утворення шестиатомного спирту
сорбітолу з глюкози за схемою
Таким чином, фруктокіназний шлях обміну
фруктози перетинається з гліколізом
на етапі утворення двох молекул
фосфотріози — гліцеральдегід-3-фосфату.
Оскільки перетворення фруктози на
тріози здійснюється без участі двох
регуляторних реакцій гліколізу
(гексокіназної та фосфофруктокіназної),
які обмежують швидкість процесу,
катаболізм фруктози відбувається
значно швидше порівняно з глюкозою.
Синтез ендогенної фруктози відбувається
через утворення шестиатомного спирту
сорбітолу з глюкози за схемою
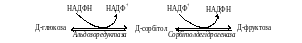 Ці
патології пов'язані з генетичними
дефектами синтезу ферментів метаболізму
фруктози. До них належать: а) непереносимість
фруктози — захворювання, молекулярною
основою якого є уроджена недостатність
фруктозо-1 -фосфатальдолази, що спричиняє
накопичення в тканинах фруктозо-1-фосфату,
який є інгібітором деяких ферментів
вуглеводного обміну, зокрема фосфорилази
глікогену. Патологія зазвичай виявляється
під час переходу від грудного годування
дітей до їжі, що містить цукор і
проявляється фруктоземією, фруктозурією
та тяжкою гіпоглікемією, яка розвиваються
після споживання продуктів, що містить
фруктозу; б) фруктоземія — патологія
обміну фруктози, спричинена недостатністю
фруктокінази. Ензимопатія характеризується
порушенням клітинної утилізації
фруктози без суттєвих клінічних проявів
Ці
патології пов'язані з генетичними
дефектами синтезу ферментів метаболізму
фруктози. До них належать: а) непереносимість
фруктози — захворювання, молекулярною
основою якого є уроджена недостатність
фруктозо-1 -фосфатальдолази, що спричиняє
накопичення в тканинах фруктозо-1-фосфату,
який є інгібітором деяких ферментів
вуглеводного обміну, зокрема фосфорилази
глікогену. Патологія зазвичай виявляється
під час переходу від грудного годування
дітей до їжі, що містить цукор і
проявляється фруктоземією, фруктозурією
та тяжкою гіпоглікемією, яка розвиваються
після споживання продуктів, що містить
фруктозу; б) фруктоземія — патологія
обміну фруктози, спричинена недостатністю
фруктокінази. Ензимопатія характеризується
порушенням клітинної утилізації
фруктози без суттєвих клінічних проявів
Метаболічний
шлях та ферментативні реакції перетворення
галактози в організмі людини. Спадкові
ензимопатії пов’язані з генетичними
дефектами синтезу ферментів метаболізму
галактози – галактоземія. Галактоза
включається в процес гліколізу складнішим
порівняно з фруктозою шляхом Специфічна
галактокіназа каталізує фосфорилування
галактози на галактозо-1-фосфат. У
наступній реакції бере участь УДФ-глюкоза,
яка використовується також для синтезу
глікогену. Фермент печінки
галактозо-1-фосфат-уридилтрансфераза
каталізує обмінну реакцію між
галактозо-1-фосфатом і УДФ-глюкозою.
Далі залишок галактози в УДФ-галактозі
під дією УДФгалактозо-4-епімерази
переходить на залишок глюкози (реакція
епімеризації біля 4-го атому вуглецю).
УДФ-глюкоза може знову взаємодіяти з
галактозо-1-фосфатом або перетворюватися
на глюкозо-1-фосфат, чи використовуватися
на синтез глікогену.
Специфічна
галактокіназа каталізує фосфорилування
галактози на галактозо-1-фосфат. У
наступній реакції бере участь УДФ-глюкоза,
яка використовується також для синтезу
глікогену. Фермент печінки
галактозо-1-фосфат-уридилтрансфераза
каталізує обмінну реакцію між
галактозо-1-фосфатом і УДФ-глюкозою.
Далі залишок галактози в УДФ-галактозі
під дією УДФгалактозо-4-епімерази
переходить на залишок глюкози (реакція
епімеризації біля 4-го атому вуглецю).
УДФ-глюкоза може знову взаємодіяти з
галактозо-1-фосфатом або перетворюватися
на глюкозо-1-фосфат, чи використовуватися
на синтез глікогену.
 У
молочній залозі УДФ-галактоза
використовується як донор галактози
в реакціях синтезу лактози, глікопротеїнів
і гліколіпідів, протеогліканів. Спадкові
ферментопатії метаболізму галактози.
Спадковий дефект синтезу
галактозо-1-фосфатуридилтрансферази
проявляється природженою ензимопатією
— галактоземією. Внаслідок неспроможності
біохімічних систем організму перетворювати
галактозу на глюкозу, у крові та
внутрішніх органах хворих нагромаджується
галактоза та галактозо-1-фосфат,
спостерігається тенденція до гіпоглікемії,
спостерігається компенсаторна
мобілізація жирів, все це клінічно
проявляється гепатомегалією, цирозом
печінки, порушенням функцій нирок,
помутнінням кришталика, затримкою
розумового розвитку.
Патологію
виявляють у ранньому дитячому віці при
споживанні молока, її розвиток може
бути сповільнений дотриманням певної
дієти (виключення з харчового раціону
продуктів, що містять галактозу).
Дуже
рідко, але все ж зустрічаються дефекти
галактокінази та уридилфосфат-4-епімерази.
Вони проявляються галактоземією,
галактозурією, проте тяжкі клінічні
прояви при цьому відсутні.
У
молочній залозі УДФ-галактоза
використовується як донор галактози
в реакціях синтезу лактози, глікопротеїнів
і гліколіпідів, протеогліканів. Спадкові
ферментопатії метаболізму галактози.
Спадковий дефект синтезу
галактозо-1-фосфатуридилтрансферази
проявляється природженою ензимопатією
— галактоземією. Внаслідок неспроможності
біохімічних систем організму перетворювати
галактозу на глюкозу, у крові та
внутрішніх органах хворих нагромаджується
галактоза та галактозо-1-фосфат,
спостерігається тенденція до гіпоглікемії,
спостерігається компенсаторна
мобілізація жирів, все це клінічно
проявляється гепатомегалією, цирозом
печінки, порушенням функцій нирок,
помутнінням кришталика, затримкою
розумового розвитку.
Патологію
виявляють у ранньому дитячому віці при
споживанні молока, її розвиток може
бути сповільнений дотриманням певної
дієти (виключення з харчового раціону
продуктів, що містять галактозу).
Дуже
рідко, але все ж зустрічаються дефекти
галактокінази та уридилфосфат-4-епімерази.
Вони проявляються галактоземією,
галактозурією, проте тяжкі клінічні
прояви при цьому відсутні.
Глюконеогенез: фізіологічне значення, ферментативні реакції, регуляторні ферменти. Метаболічний шлях глюконеогенезу: субстрати глюконеогенезу, компартменталізація перетворення пірувату в фосфоенолпіруват. Лактат та аланін як субстрати глюконеогенезу. Головном джерелом глюкози як метаболічного палива для організму людини є її надходження з харчовими продуктами у вигляді полісахариду крохмалю та сахарози. Надлишок глюкози, шо не використовується в окислювальних реакціях аеробного та анаеробного гліколізу, відкладається у вигляді енергетичних депо — глікогену та триацилгліцеролів. Втім, існують метаболічні шляхи, що забезпе- чують організм глюкозою за рахунок її синтезу з невуглеводних біомолекул. Глюконеогенез (гліконеогенез) — синтез глюкози з невуглеводних метаболіч- них попередників, до яких належать: піруват (та лактат); деякі амінокислоти (глюкогенні амінокислоти); певна кількість глюкози може утворюватися з гліце- ролу та продуктів катаболізму жирних кислот з непарною кількістю вуглецевих атомів у вуглеводневому ланцюзі. Фізіологічне значення глюконеогенезу Реакції глюконеогенезу відбуваються переважно в печінці та, в деякій мірі, в кірковому шарі нирок, оскільки в клітинах саме цих органів присутній повний набір необхідних ферментів. За добу в організмі дорослої людини синтезується до 80 г глюкози. Біосинтез глюкози забезпечує її нормальну концентрацію в умовах зменшеного надходження моносахариду із зовнішнього середовища та вичерпання головного акумульова- ного джерела глюкози — глікогену печінки та м’язів. Така фізіологічна ситуація спостерігається через декілька годин після прийому їжі (постабсорбтивний стан, що відбувається зранку, натщесерце), в умовах тривалого голодування, після вис- нажливої фізичної роботи. Особливо чутливий до зменшення внутрішньоклітин- ної концентрації глюкози головний мозок людини, для якого глюкоза є основним субстратоменергетичного обміну, що споживається в кількості близько 120 г за добу. Метаболічний шлях глюконеогенезу Метаболічний шлях глюконеогенезу є значною мірою оберненням гліколізу (що утворює з глюкози піруват та лактат) за виключенням трьох гліколітичних “кіназних” реакцій, які є термодинамічно незворотними і потребують обхідних (шунтових) механізмів. Незворотними реакціями гліколізу є: 1) гексокіназна (або глюкокіназна) реакція: глюкоза + АТФ -> глюкозо-6-фосфат + АДФ 2) фосфофруктокіназна реакція: фруктозо-6-фосфат + АТФ -> фруктозо-1,6-дифосфат + АД 3) піруваткіназна реакція: фосфоенолпіруват + АДФ -> піруват + АТФ


Глюкозо-лактатний (цикл Корі) та глюкозо-аланіновий цикли. У період відновлення після напруженої роботи молочна кислота переноситься кров'ю з м'язів до печінки, де під дією лактатдегідрогенази окиснюється до пірувату. Частина останнього використовується для глюконеогенезу, а частина розпадається аеробним шляхом, забезпечуючи процес глюконеогенезу енергією АТФ. Глюкоза потрапляє назад у скелетні м'язи і використовується для відновлення запасу глікогену. Поєднання процесу анаеробного гліколізу в скелетних м'язах і глюконеогенезу в печінці називається циклом Корі (глюкозо-лактатним циклом) При тривалому голодуванні зростає секреція корою надниркових залоз глюкокортикоїдів, які посилюють у печінці синтез ферментів глюконеогенезу (фосфоенолпіруваткарбоксикінази, глюкозо-6-фосфатази) й амінотрансфераз – ферментів, які каталізують перетворення глікогенних амінокислот на піруват і оксалоацетат. Важливим субстратом у печінці є аланін, який може утворюватися у скелетних м'язах у зворотній реакції трансамінування пірувату з глутаматом ((Піруват + Глутамат Аланін + -Кетоглутарат) Аланін, утворений у м'язах під час їх роботи, потрапляє в кров, а потім поглинається гепатоцитами (після перетворення на піруват) і використовується в глюконеогенезі.

Розщеплення та біосинтез глікогену: ферментативні реакції глікогенезу та глікогенолізу; Глікоген — тваринний гомополісахарид, що міститься в цитозолі клітин у вигляді мікроскопічних гранул і являє собою депо лабільного метаболічного палива. Глікоген є резервною формою глюкози, збереження надлишків якої у вигляді молекул мономерів неможливе у зв’язку зїї високою осмотичною актив- ністю. Основними органами, в яких депонується найбільша кількість глікогену, є печінка та скелетні м’язи, які можуть забезпечувати власні енергетичні потреби та рівень глікемії (печінка) в перервах між споживанням їжі. Глікокон’югати — складні молекули, що містять оліго- або полісахаридні ланцюги, сполученіз пептидним або ліпідним фрагментом. Нормальний біосин- тез та розщеплення вуглеводної частини глікокон’югатів є важливою передумовою реалізацїї біологічних функцій протеогліканів, глікопротеїнів та гліколіпідів.
 2.
утворення нерозгалужених ланцюгів
глікогену
2.
утворення нерозгалужених ланцюгів
глікогену
![]() Ферментативні
реакції розщеплення глікогену
(глікогенолізу). 1.
Процес глікогенолізу реалізується за
механізмом фосфоролітичного роз-
щеплення
Ферментативні
реакції розщеплення глікогену
(глікогенолізу). 1.
Процес глікогенолізу реалізується за
механізмом фосфоролітичного роз-
щеплення
 2.
Фосфорилаза відрізає моносахариднізалишки
у вигляді глюкозо-1-фосфату віднерозгалужених
амілозних ланцюгів глікогену. Розщеплення
розгалуженихфраг- ментів відбувається
під дієюаміло-1,6-глікозидази
(дерозгалужуючий фермент). Фермент
каталізує глікозилтрансферазну та
аміло-1,6-глюкозидазну реакції, а саме:
– переносить олігосахаридні залишки,
що складаються із трьох моносаха- ридів,
в кінець нерозгалужених ланцюгів, що
експонує залишки глюкози, сполучені з
основним ланцюгом α(1
6)-глікозидними зв’язками; – розриває
(1 6)-глікозидні зв’язки гідролітичним
шляхом, вивільняючи молекули глюкози.
3. Глюкозо-1-фосфат, що утворюється при
фосфоролізі глікогену, під дією
фосфоглюкомутази перетворюється в
глюкозо-6- фосфат. 4. Подальшіметаболічні
шляхи обміну глюкозо- 6-фосфату відмінні
в клітинах печінки і м’язів: – в печінці
глюкозо-6-фосфат при дії глюкозо-
6-фосфатази перетворюється у вільну
глюкозу, яка надходить у кров і
використовується в інших орга- нах і
тканинах; – у м’язах, які не містять
глюкозо-6-фосфатази, глюкозо-6-фосфат
використовується для власних енергетичних
потреб, окислюючись аеробним або
анаеробним шляхом.
2.
Фосфорилаза відрізає моносахариднізалишки
у вигляді глюкозо-1-фосфату віднерозгалужених
амілозних ланцюгів глікогену. Розщеплення
розгалуженихфраг- ментів відбувається
під дієюаміло-1,6-глікозидази
(дерозгалужуючий фермент). Фермент
каталізує глікозилтрансферазну та
аміло-1,6-глюкозидазну реакції, а саме:
– переносить олігосахаридні залишки,
що складаються із трьох моносаха- ридів,
в кінець нерозгалужених ланцюгів, що
експонує залишки глюкози, сполучені з
основним ланцюгом α(1
6)-глікозидними зв’язками; – розриває
(1 6)-глікозидні зв’язки гідролітичним
шляхом, вивільняючи молекули глюкози.
3. Глюкозо-1-фосфат, що утворюється при
фосфоролізі глікогену, під дією
фосфоглюкомутази перетворюється в
глюкозо-6- фосфат. 4. Подальшіметаболічні
шляхи обміну глюкозо- 6-фосфату відмінні
в клітинах печінки і м’язів: – в печінці
глюкозо-6-фосфат при дії глюкозо-
6-фосфатази перетворюється у вільну
глюкозу, яка надходить у кров і
використовується в інших орга- нах і
тканинах; – у м’язах, які не містять
глюкозо-6-фосфатази, глюкозо-6-фосфат
використовується для власних енергетичних
потреб, окислюючись аеробним або
анаеробним шляхом.

Каскадні механізми цАМФ-залежної регуляції активностей глікоген фосфорилази та глікогенсинтази. Глікогенфосфорилаза печінки там’язів — це димер, якиймістить у своєму складі піридоксальфосфат. Фермент може перебувати в активній фосфорильованій формі (фосфорилаза а) та неактивній дефосфорильованій формі (фосфорилаза b). 1. Перетвореннянеактивноїфосфорилази b в активнуфосфорилазу a відбувається шляхом фосфорилювання серинових залишків у молекулі фосфорилази за рахунок макроергічних зв’язків АТФ: фосфорилаза b + 4АТФ фосфорилаза a + 4АДФ (димер) (тетрамер) Дефосфорилювання фосфорилази a протеїнфосфатазою (фосфатазою) призводить до утворення неактивної фосфорилази b. 2. Фермент, що фосфорилює фосфорилазу b, — кіназа фосфорилази b. Кіназа фосфорилази b також існує в двох молекулярних формах — неактивній та активній, які взаємоперетворюються шляхом фосфорилювання — дефосфорилювання. Фосфорилювання кінази фосфорилази b (КФ b) відбувається за участю АТФ під дією цАМФ-залежної протеїнкінази: цАМФ-залежна протеїнкіназа КФ b (неактивна) + АТФ --->КФ b (активна) + АДФ Дефосфорилювання кіназифосфорилазиb фосфатазоюсупроводжується зво- ротним переходом ферменту в неактивну форму. 3. Каталітично активна протеїнкіназа формується за умов її взаємодії з 3',5'-АМФ (цАМФ), утворення якого в аденілатциклазній реакції є пусковою молекулярною подією, що включає каскадний механізм регуляції ферментних процесів у клітині (глава 7). 4. У м’язах існують також механізми стимуляції глікогенолізу шляхом ало- стеричної активації фосфорилази b та кінази фосфорилази b без фосфорилю- вання цих білків ферментного каскаду: – до складу кінази фосфорилази b входить субодиниця, що є Са2+-зв’язуючим білком, за своїми властивостями ідентичним кальмодуліну. Зв’язування цією субодиницею чотирьох іонів кальцію активує каталітичний центр кінази фосфо- рилази b, хоча молекула ферменту залишається в дефосфорильованому стані. Цей процес має фізіологічне значення термінового механізму забезпечення енер- гією м’язового скорочення, який включається в результаті вивільнення Са2+ з саркоплазматичного ретикулума після надходження нервового імпульсу; – активація м’язової фосфорилази b м’язів може також відбуватися без її фосфорилювання. Алостеричним активатором ферменту є АМФ, концентрація якого в м’язах значно зростає в умовах тривалого фізичного навантаження, що призводить до вичерпання резервів АТФі накопичення АДФта АМФ. Активована АМФ фосфорилаза b здатна забезпечити швидкість глікогенолізу, достатню для виконання м’язової роботи помірної інтенсивності.

Гормональна регуляція обміну глікогену в м'язах та печінці. Участь гормонів у регуляції глікогенолізу та глікогенезу в м’язах та печінці може бути підсумована таким чином: А. У м’язах. Адреналін — стимулює глікогеноліз та гальмує глікогенез шляхом: а) активації глікогенфосфорилази за рахунок її цАМФ-залежного фосфори- лювання; б) інгібірування глікогенсинтази за рахунок її цАМФ-залежного фосфори- лювання. Інсулін — стимулює глікогенез і гальмує глікогеноліз шляхом: а) підвищення проникності мембран м’язових клітин для глюкози, що вико- ристовується для синтезу глікогену; б) зменшення внутрішньоклітинного рівня цАМФ за рахунок активації її розщеплення фосфодіестеразою. Б. У печінці. Глюкагон — стимулює глікогеноліз та гальмує глікогенез за механізмом, аналогічним дії адреналізу в клітинах м’язів. Інсулін — підвищує активність ферментативних реакцій синтезу глікогену за рахунок біохімічних механізмів, близьких до розглянутих вище. Таким чином, співвідношення глюкагон/інсулін є важливим фізіологічним механізмом, що контролює глікогенну функцію печінки та рівень глюкози в крові після споживання їжі: – переважання інсуліну сприяє утворенню в організмі резервів вуглеводів у формі глікогену печінки; – переважання глюкагонусприяє мобілізаціїзапасів глікогену печінки в умовах зниження рівня глюкоземії, яке спостерігається через кілька годин після спожи- вання їжі.
Генетичні порушення ферментів метаболізму глікогену: глікогенози – аномально високе накопичення глікогену в органах і тканинах, аглікогенози – недостатне запасання глікогену в тканинах. Спадкові порушення реакцій розщеплення або синтезу глікогену проявляються у вигляді ензимопатій — хвороб накопичення глікогену — глікогенозів та агліко- генозів. Глікогенози Глікогенози — спадкові хвороби, молекулярною основою виникнення яких є уроджена недостатність синтезу певних ферментів глікогенолізу, пов’язана з де- фектами в генетичній системі клітин. При глікогенозах у внутрішніх органах та тканинах (здебільшого в печінці, м’язах, клітинах крові) спостерігається нако- пичення аномально надмірної кількості глікогену, інодізізміненою молекулярною структурою, який не може використовуватися у метаболічних процесах — таб- лиця 13.2. Т а б л и ц я 13.2. Біохімічні характеристики глікогенозів (за R.Berkow (Ed.): The Merck Manual of Diagnosis and Therapy, 1992, іззмінами) Клінічно глікогенози проявляються важкою гіпоглюкоземією внаслідок не- здатності глікогену печінки розщеплюватися з вивільненням молекул глюкози. Глікогенози, при яких ушкоджені ферментні системи мобілізації глікогену печін- ки, характеризуються збільшенняммаси органа, жировою дистрофією гепатоцитів та явищами цирозу. Недостатність ферментних систем глікогенолізу в м’язах су- проводжується судомами за умов фізичних навантажень. Аглікогенози Аглікогенози — спадкові хвороби накопичення глікогену, молекулярною основою яких є генетичні дефекти, що призводять до порушення утворення фер- менту глікогенсинтази. Внаслідок недостатності глікогенсинтази гепатоцити не здатні утворювати резерви глікогену, концентрація якого всередині клітин значно зменшена. Внас- лідок відсутності глікогенових резервів хворі при аглікогенозах, як і при глікоге- нозах, страждають від глибокої гіпоглюкоземії, особливо натщесерце, після знач- ної перерви з часу надходження харчової глюкози. Гіпоглюкоземія при аглікогено- зах може супроводжуватися важкою комою внаслідок енергетичного голодування головного мозку. Такі хворі звичайно вмирають у ранньому дитячому віці.
Метаболізм
вуглеводних компонентів глікокон'югатів.
Глікозилювання
невуглеводних молекул є важливим класом
біохімічних реак- цій, що призводять
до утворення гібридних молекул
глікокон’югатів. У результаті реакцій
глікозилювання олігосахаридні та
полісахаридні ланцюги приєднуються
ковалентними зв’язками до поліпептидів
або ліпідів, утворюючи фізіологічно
важливі глікопротеїни, гліколіпіди та
протеоглікани. Біосинтез глікон’югатів
Процеси глікозилювання білків і ліпідів
відбуваються в ендоплазматичному
ретикулумі та апараті Гольджі і
каталізуються специфічними
глікозилтрансфера- зами. Біохімічні
реакції синтезу олігосахаридних
фрагментів глікопротеїнів, глі- коліпідів
та гетерополісахаридних фрагментів
протеогліканів (глікозамінгліканів)
в цілому співпадають і будуть розглянуті
на прикладі більш детального вивченого
процесу утворення вуглеводних компонентів
глікопротеїнів. Проте існують суттєві
відмінності вмолекулярнихмеханізмах
біосинтезуО- та N-зв’язаних
глікопротеїнів. 1. Синтез О-зв’язаних
глікопротеїнів Олігосахаридні ланцюги
глікопротеїнів О-глікозидного типу
конструюються шляхом ступеневого
приєднання моносахаридних залишків
до ОН-груп серину або треоніну
поліпептидної частини молекули. Донорами
вуглеводних залишків у цих реакціях є
нуклеотидцукри, зокрема
УДФ-N-ацетилгалактозамін
(UDPGalNAc),
УДФ-галактоза (UDP-Gal)
та ЦМФ-N-ацетилнейрамінова
(сіалова) кис- лота (СMP-NeuAc).
Ферменти, що каталізують перенос
моносахаридного залишку від нуклеотид-
цукрів на OH-Ser(Thr),
— мембранозв’язані
глікопротеїн-глікозилтрансферази. 2.
Синтез N-зв’язаних
глікопротеїнів Особливістю синтезу
N-зв’язаних
глікопротеїнів є участь високомолекулярного
спирту ізопреноїдної природи —
доліхолфосфату, який виступає в ролі
проміж- ного переносника олігосахаридних
фрагментів. За хімічною будовою доліхол
є високомолекулярним спиртом, що,
подібно до каучука, має найбільшу
довжину з природних вуглеводнів.
Гідрофобна основа доліхолу занурена
в товщу ліпідного біслою біомембран,
а ОН-група, що взаємодіє з водною фазою,
здатна акцептувати моносахаридні
фрагменти при послідовній дії специфічних
глікозилтрансфераз.
 3.
Синтез гліколіпідів Гліколіпіди —
важливі структурні компоненти біомембран,
що визначають, разом із мембранними
глікопротеїнами, антигенні властивості
клітин. Подібно до утворення глікопротеїнів,
формування гліколіпідів, зокрема гліко-
сфінголіпідів, відбувається за механізмом
послідовного нарощування олігосаха-
ридів за участю специфічних
глікозилтрансфераз. Донорами моносахаридів
та їх похідних є нуклеотидцукри, що
переносять залишки цукрів на молекулярну
основу, якою виступає церамід(ацилсфінгозин).
Процес відбувається за схемою:
Катаболізм
глікокон’югатів Розщеплення
гетерополісахаридних фрагментів
глікокон’югатів — протео- гліканів,
глікопротеїнів і гліколіпідів
реалізується за участю глікозидаз, що
локалізовані в лізосомах, та специфічні
до різних типів глікозидних зв’язків.
Найбільш вивченими є лізосомальні
ферменти, які беруть участь у деградації
вуглеводних компонентів протеогліканів
(глікозамінгліканів) та гліколіпідів.
Уроджені порушення розщеплення цих
сполук (ензимопатії) проявляються
гліко- зидозами, за яких відбувається
внутрішньоклітинне накопичення певних
форм гетерополісахаридів. Деградація
гетерополісахаридних структур
протеогліканів каталізується фер-
ментами, які за своєю субстратною
специфічністю поділяються на ендоглікози-
дази, екзоглікозидази, сульфатази. До
глікозидаз, що беруть участь у катаболізмі
глікозамінгліканів, належать:
Гіалуронідаза — широко розповсюджена
в тканинах ендоглікозидаза, що діє на
гіалуронову кислоту та хондроїтинсульфати,
розщеплюючи їх до тетра- сахаридних
фрагментів. β-Глюкуронідаза
— екзоглікозидаза, що відщеплює
глюкуронову та ідуронову кислоту від
тетрасахаридів (див. вище) та таких
гетерополісахаридів, як дерма-
тансульфати, гепарансульфати,
хондроїтинсульфати, гіалуронова
кислота. При
спадковій недостатності ферменту у
хворих спостерігається виділення
глікозамін- гліканів із сечею.
β-Галактозидази — множинні ферменти,
що розщеплюють внутрішні β-га- лактозидні
зв’язки в олігосахаридних залишках
протеогліканів, глікопротеїнів та
гліколіпідів. β-D-Ацетилгексозамінідаза
— екзоглікозидаза, що відщеплює від
вуглеводних компонентів глікокон’югатів
термінальні N-ацетилгексозаміни GlcNAc
та GalNAc. Спадкова недостатність ферменту
спостерігається при таких гліколіпідозах,
як хвороби Тея-Сакса та Зандхоффа.
α-L-Ідуронідаза — екзоглікозидаза, що
відщеплює від полісахаридних ланцю-
гів термінальні залишки L-ідуронової
кислоти. Відомо декілька форм спадкових
порушень, пов’язаних із недостатністю
цього ферменту. Існує декілька
лізосомальних сульфатаз, що відщеплюють
сульфатнізалишки від різних мономерних
одиниць у складі глікозамінгліканів,
зокрема арилсульфа- таза А, арилсульфатаза
В та арилсульфатаза С. Недостатність
сульфатаз проявляється декількома
формами глікозидозів.
3.
Синтез гліколіпідів Гліколіпіди —
важливі структурні компоненти біомембран,
що визначають, разом із мембранними
глікопротеїнами, антигенні властивості
клітин. Подібно до утворення глікопротеїнів,
формування гліколіпідів, зокрема гліко-
сфінголіпідів, відбувається за механізмом
послідовного нарощування олігосаха-
ридів за участю специфічних
глікозилтрансфераз. Донорами моносахаридів
та їх похідних є нуклеотидцукри, що
переносять залишки цукрів на молекулярну
основу, якою виступає церамід(ацилсфінгозин).
Процес відбувається за схемою:
Катаболізм
глікокон’югатів Розщеплення
гетерополісахаридних фрагментів
глікокон’югатів — протео- гліканів,
глікопротеїнів і гліколіпідів
реалізується за участю глікозидаз, що
локалізовані в лізосомах, та специфічні
до різних типів глікозидних зв’язків.
Найбільш вивченими є лізосомальні
ферменти, які беруть участь у деградації
вуглеводних компонентів протеогліканів
(глікозамінгліканів) та гліколіпідів.
Уроджені порушення розщеплення цих
сполук (ензимопатії) проявляються
гліко- зидозами, за яких відбувається
внутрішньоклітинне накопичення певних
форм гетерополісахаридів. Деградація
гетерополісахаридних структур
протеогліканів каталізується фер-
ментами, які за своєю субстратною
специфічністю поділяються на ендоглікози-
дази, екзоглікозидази, сульфатази. До
глікозидаз, що беруть участь у катаболізмі
глікозамінгліканів, належать:
Гіалуронідаза — широко розповсюджена
в тканинах ендоглікозидаза, що діє на
гіалуронову кислоту та хондроїтинсульфати,
розщеплюючи їх до тетра- сахаридних
фрагментів. β-Глюкуронідаза
— екзоглікозидаза, що відщеплює
глюкуронову та ідуронову кислоту від
тетрасахаридів (див. вище) та таких
гетерополісахаридів, як дерма-
тансульфати, гепарансульфати,
хондроїтинсульфати, гіалуронова
кислота. При
спадковій недостатності ферменту у
хворих спостерігається виділення
глікозамін- гліканів із сечею.
β-Галактозидази — множинні ферменти,
що розщеплюють внутрішні β-га- лактозидні
зв’язки в олігосахаридних залишках
протеогліканів, глікопротеїнів та
гліколіпідів. β-D-Ацетилгексозамінідаза
— екзоглікозидаза, що відщеплює від
вуглеводних компонентів глікокон’югатів
термінальні N-ацетилгексозаміни GlcNAc
та GalNAc. Спадкова недостатність ферменту
спостерігається при таких гліколіпідозах,
як хвороби Тея-Сакса та Зандхоффа.
α-L-Ідуронідаза — екзоглікозидаза, що
відщеплює від полісахаридних ланцю-
гів термінальні залишки L-ідуронової
кислоти. Відомо декілька форм спадкових
порушень, пов’язаних із недостатністю
цього ферменту. Існує декілька
лізосомальних сульфатаз, що відщеплюють
сульфатнізалишки від різних мономерних
одиниць у складі глікозамінгліканів,
зокрема арилсульфа- таза А, арилсульфатаза
В та арилсульфатаза С. Недостатність
сульфатаз проявляється декількома
формами глікозидозів.
Генетичні порушення метаболізму глікокон'югатів (глікозидози). Глікозидози — генетичні порушення метаболізму гетеполісахаридів, що є структурними компонентами протеогліканів та гліколіпідів. Глікозидози, які пов’язані із спадковою недостатністю ферментів розщеплення вуглеводних компонентів протеогліканів, мають назву мукополісахаридозів, гліколіпідів — гліколіпідозів (або муколіпідозів і сфінголіпідозів). Глікозидози і, зокрема, мукополісахаридози, є тяжкими захворюваннями, які проявляються глибокими порушеннями з боку сполучної тканини багатьох внутрішніх органів, зокрема, помутнінням рогівки, патологією кісток та суглобів, затримкою розвитку дитини та скороченням тривалості життя. Діагностичне значення має надмірна екскреція певних глікозамінгліканів із сечею. Біохімічні характеристики найбільш розповсюджених мукополісахаридозів (МПС) людини, тобто спадкових порушень катаболізму глікозамінгліканів.
Гормони – регулятори обміну глюкози (глюкагон, адреналін, глюкокортикоїди, соматотропін, інсулін - ефекти та механізми впливу на рівень глюкоземії). 2. Гормональна регуляція глюконеогенезу — здійснюється за участю глю- кагону, адреналіну (епінефрину), глюкокортикоїдних гормонів кори надниркових залоз та інсуліну. 2.1. Глюкагон, адреналін та глюкокортикоїди підвищують швидкості синтезу в гепатоцитах шунтових ферментів глюконеогенезу — ФЕП-кінази, Фр-1,6- дифосфатази, Г-6-Ф-ази. 2.1. Інсулін пригнічує синтез зазначених глюконеогенних ферментів, що гальмує активність процесу глюконеогенезу
Глюкоземія: нормальний стан та його порушення (гіпер-, гіпоглюкоземія та глюкозурія). Співвідношення глюкагон/інсулін має найбільше значення для регуляції взаємовідношення між активностями процесів глюконеогенезу та гліколізу. 1. Зменшення рівня глюкоземії(гіпоглюкоземія, гіпоглікемія), що настає че- рез декілька годин після останнього споживання їжі, супроводжується підви- щенням рівня секреції α-клітинами острівкової частини підшлункової залози гормонуглюкагону, який стимулює процеси глюконеогенезу за розглянутими вище механізмами. За рахунок активації каскадної аденілатциклазної системи в мемб- ранах гепатоцитів глюкагон стимулює фосфороліз глікогену, що також робить свій внесок у збільшення рівня вільної глюкози. 2. Збільшення рівня глюкоземії (гіперглюкоземія, гіперглікемія) стимулює секреціюβ-клітинами підшлунковоїзалози гормонуінсуліну, який підвищує ступінь проникності плазматичнихмембран багатьох клітин для глюкози (окрімголовного мозку), сприяючи їх внутрішньоклітинному метаболізму. Інсулін зменшує швид- кість синтезу ферментів глюконеогенезу в печінці та, навпаки, стимулює синтез ключових регуляторних ферментів гліколізу — гексокінази, фосфофруктокінази, піруваткінази, переводячи, таким чином, обмін глюкози з глюкогенного на гліко- літичний шлях. Крім того, інсулін стимулює синтез у печінці та м’язах глікогену, що також є метаболічним процесом, спрямованим на зменшення концентрації вільної глюкози. Адреналін—гормонмозкової частини надниркових залоз, збільшення секреції якого призводить до підвищення вмісту глюкози в крові за рахунок стимуляції фосфоролізу глікогену (детально – див. главу 13) в м’язах та частково в печінці, що, через Г-6-Ф-азну реакцію, супроводжується гіперглюкоземією. Глюкокортикоїди, основним представником яких є кортизол, стимулюють глюконеогенез (і, відповідно, підвищують рівень глюкоземії при тривалому вве- денні в організм) за рахунок стимуляції синтезу в печінці ферментів глюконео- генезу — головним чином ФЕП-кінази, та ферментів, що перетворюють у субст- рати глюконеогенезу деякі глюкогенні амінокислоти (серин, тирозин, триптофан). Соматотропін — гормон аденогіпофіза, що, подібно до інсуліну, збільшує проникність плазматичних мембран клітин м’язової та жирової тканини для глюкози, але, на відміну від інсуліну, — активує глюконеогенез в печінці.
Цукровий діабет; інсулінозалежна та інсулінонезалежна форми. Цукровий діабет — ендокринна хвороба, що характеризується генетично детермінованимабсолютнимабовідноснимдефіцитомгормонупідшлунковоїзалози інсуліну. Воснові розвитку цукрового діабету лежить зниження продукції інсуліну в β-клітинах острівкового (інсулярного) апарату підшлункової залози або нездатність відповідних клітинних рецепторів реагувати на інсулін. Відповідно до зазначеного розрізняють: – інсулінозалежний цукровий діабет (ІЗЦД), який розвивається внаслідок руйнування значної кількості (звичайно більше 90 %) секретуючих інсулін β-клі- тин. Причиною деструкціїβ-клітин є генетично зумовлений автоімунний процес. ІЗЦД складає 10-15 % всіх випадків цукрового діабету і проявляється гіпер- глікемією та схильністю до кетонемії та кетоацидозу. Ця форма цукрового діабету розвивається звичайно в ранньому віці (до 30 років), найчастіше у дітей та підлітків; – інсулінонезалежний цукровий діабет (ІНЗЦД) — форма цукрового діабету, за якого у більшості хворих зберігаються β-клітини в інсулярній частині підшлункової залози, але порушені специфічні реакції клітин на дію інсуліну або регуляція його секреції під впливом збільшеної концентрації глюкози крові. ІНЗЦД розвивається звичайно у дорослих (старше 30 років) та осіб похилого віку і проявляється гіперглікемією та ожирінням. Найбільш характерним клініко-біохімічним проявом цукрового діабету є збіль- шення (порівняно з нормою) рівня глюкози в крові в умовах натщесерце або після прийому їжі, який перевищує значення, характерні дляфізіологічної, алімен- тарної гіперглюкоземії, досягаючи значень 500 мг % та більше. При легких фор- мах захворювання гіперглюкоземіяне спостерігається в постабсорбтивному стані і виявляється лише за умов визначення толерантності організму до глюкози, яке здійснюється шляхом “цукрового навантаження”. Стандартним методом виявлення цукрового діабету є “оральний глюкозотоле- рантний тест”, що здійснюється шляхом надання пацієнту (після 10-14-годинного голодування) per os розчину, що містить 75 г глюкози; контрольні визначення глю- кози крові проводять протягом 2 год з 15-хвилинним інтервалом.
Клініко-біохімічна характеристика та діагностичні критерії цукрового діабету. Глюкозотолерантний тест, подвійне цукрове навантаження. Крім зазначеного методу з одноразовим введенням пацієнту глюкози, викори- стовують також “подвійні цукрові навантаження” (проба Штауба-Трауготта) (рис. 12.8). У разі зростання рівнів глюкоземії більше 180 мг % (“нирковий поріг” для глюкози), остання починається виводитися із сечею — глюкозурія. Важкі форми некомпенсованого цукрового діабету супроводжуються значними зрушеннями ліпідного обміну — кетонемією та кетонурією , а також підви- щеним катаболізмом білків і амінокислот із розвитком азотемії та азотурі
Адипоцити жирової тканини та їх роль в обміні ліпідів і біоенергетичних процесах в організмі.
Основне місце локалізації резервних тригліцери- дів в організмі людини — адипоцитижирової тканини (ліпоцити), значна частина цитоплазми яких зайнята гігантськоюсферичною ліпідноюкраплею — рис.14.1. У значно меншій кількості тригліцериди наявні в клітинах інших органів, зокрема гепатоцитів печінки, де вони перебувають у виглядіжирових краплин у ци- топлазмі. Жирова тканина, до 65 % маси якої припадає на нейтральніжири, є високоспеціалізованою тканиною, що акумулює значні кількості метаболічного палива, енергетична цінність якого суттєво перевищує енергетичні резерви вуглеводів і білків. Масажирової тканини у дорослої людини середньої ваги дорівнює в середньому 10 кг, ізагальний вміст тригліцеридів у ній достатній для забезпечення енергетичних потреб організму протягом, принаймні, 40 днів голодування
Катаболізм триацилгліцеролів: реакції; механізми регуляції активності тригліцеридліпази. Нейрогуморальна регуляція ліполізу за участю адреналіну, норадреналіну, глюкагону, інсуліну. Триацилгліцероли (нейтральні жири, жири) потрапляють до організму людини як компоненти тваринної і рослинної їжі. Ліпіди цього класу розщеплюються в травному каналі до моногліцеридів, вільнихжирних кислот та гліцеролу, які після всмоктування в кишечнику та складних біохімічних перетворень в ентероцитах, крові та печінці депонуються вжировій тканині. Накопичення резервів нейтраль- нихжирів є найбільш ефективним механізмом акумулюванняметаболічної енер- гії, оскільки завдяки високому ступеню відновлення жирнокислотних залишків, що входять до складу молекул триацилгліцеролів, при їх окисленні вивільняється значно більше хімічної енергії, ніж при катаболізмі вуглеводів та білків. Зазначений ступеневий процес ліполізу, що відбувається в адипоцитах жи- рової тканини, каталізується трьома ферментами — тригліцерид-, дигліцерид- та моногліцеридліпазою. Активність двох останніх ферментів (E2 та E3 ) в де- кілька десятків разів перевищує активність першого ферменту (E1 ). Звичайно загальна швидкість багатоступеневого метаболічного ланцюга контролюється активністю ферменту, що каталізує найбільш повільну (лімітуючу) стадію про- цесу. Тому такий фермент єрегуляторним, і, дісно, активність тканинноїтриглі- церидліпази (ТГ-ліпази) регуляється багатьма гормонами, зокрема адреналіном, глюкагоном, інсуліном, соматотропіном. Молекулярної основою регуляції активності тригліцеридліпази адипоци- тів є її ковалентна модифікація шляхом оберненого фосфорилювання — дефосфорилювання. Адреналін та норадреналін — катехоламіни, що активують ліполіз у жировій тканиніза рахунок стимуляції цАМФ-залежного каскадного механізму регуляції активності ТГ-ліпази адипоцитів. Ліполітична дія цих гормонів реалізується за умов фізіологічних (фізичне напруження, зниження температури навколишнього середовища) та психологічних (страх, тривога) стресів, що супроводжуються ви- вільненням з мозкового шару наднирникових залоз адреналіну, а також стиму- ляцією симпатичної нервової системи та вивільненням у синапсах нейронівнор- адреналіну, що взаємодіють із адренергічними рецепторами мембран адипоцитів. Глюкагон — панкреатичний гормон, що стимулює ліполітичну систему вжиро- вій тканиніза механізмом, подібним до дії катехоламінів, тобто за рахунок підви- щення в адипоцитах вмісту цАМФ, пов’язаного з активацією аденілатциклази. Дія глюкагону проявляється в умовах зниження концентрації глюкози в крові через зменшення її надходження з кишечника або посиленого використання в тканинах. У цілому за рахунок розглянутих біохімічних механізмів метаболічні ефекти катехоламінів та глюкагону призводять до швидкої стимуляції глікогенолізу в печінці і м’язах та ліполізу в жировій тканині, що забезпечує підвищені енерге- тичні потреби організму за умов стресу або голодування. Інсулін На відміну від зазначених гуморальних факторів, що активують ТГ-ліпазу адипоцитів, спричиняючи мобілізацію НЕЖК із жирової тканини, гормон інсулін гальмує процес ліполізу та вивільнення жирних кислот. Інгібіруюча дія інсуліну відносно ліполізу в адипоцитах реалізується за рахунок двох біохімічних меха- нізмів: а) зменшення концентрації цАМФ, що може бути пов’язаним з активацією фосфодіестерази цАМФ; б) збільшення проникності мембран адипоцитів до глюкози, результатом чого є активація в жировій тканині гліколізу і, відповідно, накопичення гліколітичних метаболітів діоксіацетонфосфату та 3-фосфогліцеринальдегіду. Ці метаболіти, в свою чергу, є попередниками гліцерол-3-фосфату, що необхідний для реетери- фікаціїжирних кислот при біосинтезі триацилгліцеролів. Таким чином, стимульо- ване інсуліном підвищене надходження в адипоцити глюкози переключає мета- болізм жирних кислот на використання їх здебільшого в синтетичних реакціях і зменшує їх вихід у кров. Соматотропін — гормон передньої частки гіпофіза, який також стимулює ліполіз у жировій тканині за умов голодування, але його ліполітична дія суттєво відрізняється від дії катехоламінів та глюкагону. Соматотропін спричиняє підви- щення процесів ліполізу за рахунок підвищення синтезу відповідних ферментних білків. Метаболічні ефекти соматотропіну розвиваються повільно, що свідчить про його значення в поступовій адаптації до голодування.
