

1. Основные направления развития фармацевтической химии и её задачи на современном этапе.
Фармацевтическая химия – прикладная наука, которая базируется на общих законах химических наук (органической, неорганический, аналитической, физической и коллоидной химии), исследует способы получения, строение, физические и химические свойства ЛВ, взаимодействие между их химической структурой и фармакологическим действием, методы контроля качества и изменения, происходящие при хранении ЛВ.
Основные методы исследования – анализ, синтез.
Задачи решаются с помощью классических физических, физико-химических методов, которые используются для синтеза, анализа ЛВ.
Основные направления ФХ:
1) создание и исследование новых ЛС.
2) разработка способов фармацевтического и биофармацевтического анализа.
Приоритетные научные направления ФХ на современном этапе: синтез эффективных БАВ химическим, микробиологическим, генно-инженерным методами; исследование БАВ, содержащихся в растительном и животном сырье и получение из него ЛС; разработка и совершенствование способов анализа ЛВ с учетом современных требований к их качеству; обоснование условий хранения и сроков годности ЛС; исследования в области стандартизации и совершенствование НД путем включения новых методик в ФС, ФСП и др.
2. Методологические основы и принципы классификации ЛС (преимущества и недостатки различных типов классификации).
Существует 2 основных типа классификации:
1) Химическая (по химической структуре) – используется специалистами, которые работают в области ФХ.
2) Фармакологическая (по характеру действия ЛВ на организм) – подходит для медицинской практики.
Химическая – позволяет четко разделить вещества по группам и классам соединений в зависимости от их химической структуры; в 1 химическую группу входят вещества с различным фармакологическим действием.
ЛВ: неорганические (расположение элементов в таблице Менделеева и по основным классам: оксиды, гидроксиды, кислоты, щелочи, соли, комплексы); органические (как в органической химии).
Органические: строение УВ (алифатические и циклические: гетероциклические (5-членные циклы…) и карбоциклические: алициклические и ароматические), строение функциональной группы (галогенпроизводные, спирты, фенолы, простые эфиры, альдегиды, кетоны и др.)
Фармакологическая – позволяет врачу сориентироваться и выбрать заменитель, агонист или антагонист ЛВ; в 1 группу попадают вещества различного химического строения.
АТХ – все ЛВ делятся на группы в зависимости от органа и системы, на которую они действуют, а также от их терапевтических характеристик и химического строения.
Фармакотерапевтическая классификация (Машковский) – имеет значение для врачей и провизоров (14 групп – классы – подклассы).
Классификация МНН (ВОЗ).
3. Получение ЛС (природные источники, химические и биологические синтезы, генная инженерия).
Органические и неорганические ЛВ могул получать синтетическим путем, либо выделением из природных источников, либо методом биотехнологии и генной инженерии.
Неорганические ЛВ – получают из минерального сырья (горные породы, минеральные источники, соляные озера и др.)
Органические ЛВ – нефть (разделение на фракции); растения (более 30% препаратов растительного происхождения, они обладают меньшим количеством побочных действий, меньшей токсичностью; могут служить для получения полусинтетических препаратов с более высоким фармакологическим эффектом; ЛРС до сих используется для получения сердечных гликозидов, алкалоидов, ДВ, терпеноидов; получение галеновых препаратов (суммарные – настойки, экстракты).
Продукты для синтеза ЛВ: каменноугольная смола, каменный уголь, фракции нефти, горючие сланцы – нефтехимическая, коксохимическая, лесохимическая промышленность.
Биотехнология – технология получения продуктов из живых клеток различного происхождения. Объекты: культивируемые ткани и клетки животных и растений, а также микроорганизмы, созданные методами генной инженерии.
Отрасли биотехнологии:
1) протеиновая технология – использование генетически измененных микроорганизмов (интерфероны – дрожжевые клетки).
2) метод моноклональных антител – создание гибридов, продуцирующих моноклональные антитела к определенным бактериям, вирусам.
3) инженерная энзимология – получение иммобилизированных ферментов (термостабильность, интервал pH, многократное использование).
Генная инженерия (клеточная инженерия) – использование клеток, либо манипуляции с этими клетками для создания новых технологий. Она осуществляет введение в клетку чужого генетического материала (фрагменты ДНК, плазмиды, хромосомы), в результате получаются клетки с новыми свойствами, продуцирующие вещества, которые не характерны для исходных клеток (инсулин, интерферон).
4. Специфические особенности фармацевтического анализа.
ФА – наука о химической характеристике и измерении БАВ на всех этапах: от контроля сырья до оценки качества ЛВ, изучение его стабильности, установление сроков годности и стандартизация ЛФ.
Особенности: специфичность, чувствительность, точность, экспрессность (мало времени и min количества испытуемого ЛВ и реактивов).
Установление подлинности (подтверждение идентичности анализируемого ЛВ) – внешний вид, физические свойства, константы, растворимость; тверды ЛФ – форма кристаллов, гигроскопичность, устойчивость к свету, кислороду, запах; жидкие ЛФ – цвет, запах, летучесть. ФА детерминирован.
Формы ФА:
1) фармакопейный анализ – с помощью ГФ.
2) постадийный контроль производства ЛС.
3) анализ ЛФ индивидуального изготовления.
4) экспресс-анализ.
5) биофармацевтический анализ = связан с фармакологией; in vitro, in vivo.
Критерии: избирательность (воспроизводимость, правильность), чувствительность (предел обнаружения), точность, экспрессность (мало времени и min количества испытуемого ЛВ и реактивов). Для количественного определения – точность, избирательность; чувствительность отбрасывают при большой навеске ЛВ.
Ошибки в ФА:
1) Грубые – неправильные расчеты (отбрасываются).
2) Систематические – отражают правильность анализа.
3) Случайные – воспроизводимость результатов.
5. Источники и причины недоброкачественности лекарственных веществ.
Процессы в ЛВ могут привести к изменению химического состояния и свойств. Эти процессы могут привести к потере фармакологической активности, к образованию примесей, имеющих иную направленность фармакологического действия или могут образовываться токсичные вещества.
Факторы:
1) физические: температура, влага, свет.
2) химические: тяжелые Ме.
3) биологические – рост микроорганизмов.
Температура – с увеличением увеличивается скорость реакции; с понижением (понижается активность MgSO4, CaCl2, раствора адреналина).
Свет – повышается скорость разложения; кристаллические сухие вещества более устойчивы, чем растворы; изменение цвета при длительном освещении; некоторые вещества сохраняют свою активность (содержащие железо, при этом повышается их стабильность).
Влага – снижает фармакологическую активность; + и – влияет на ЛВ; гигроскопичность.
Источники примесей: исходные и промежуточные продукты синтеза; сопутствующие вещества (растворители, остатки кислот, щелочей, участвующих в производстве); ионы тяжелых металлов (аппаратура).
Классификация примесей:
1) про происхождению: технологические, приобретенные.
2) по фармакологическому действию: индифферентные, токсичные.
Различают: общие примеси – могут не оказывать токсического действия; специфические – характерны для конкретного ЛВ.
6. Общие требования к испытаниям на чистоту.
Различают: общие примеси – могут не оказывать токсического действия; специфические – характерны для конкретного ЛВ.
Общие требования, которые предъявляются к испытаниям на чистоту - чувствительность, специфичность и воспроизводимость используемой реакции, а также пригодность ее применения для установления допустимых пределов содержания примесей.
Определить максимальное содержание примесей в испытуемом препарате можно двумя путями (эталонным и безэталонным). Один из них основан на сравнении с эталонным раствором (стандартом). При этом в одинаковых условиях наблюдают окраску или помутнение, возникающие под действием какого-либо реактива. Эталон представляет собой образец, содержащий определенное количество открываемой примеси. Установление наличия примесей производят колориметрическим или нефелометрическим методом, сравнивания результаты реакций в растворе эталона и в растворе препарата после добавления одинаковых количеств соответствующих реактивов. При выполнении испытаний на чистоту необходимо строго соблюдать общие указания, предусмотренные фармакопеями. Вода и используемые реактивы не должны содержать ионов, наличие которых устанавливают; одинакового диаметра и бесцветными должны быть пробирки; навески должны отвешиваться с точностью до 0,001 г; реактивы следует добавлять одновременно и в одинаковых количествах, как к эталонному, так и к испытуемому раствору; образующуюся опалесценцию наблюдают в проходящем свете на темном фоне, а окраску - в отраженном свете на белом фоне. Если устанавливают отсутствие примеси, то к испытуемому раствору прибавляют все реактивы, кроме основного; затем полученный раствор делят на две равные части и к одной из них прибавляют основной реактив. При сравнении не должно быть заметных различий между обеими частями раствора. Последовательность и скорость прибавления реактива влияют на результаты испытаний на чистоту. Иногда необходимо также соблюдать интервал времени, в течение которого следует вести наблюдение за результатом реакции.
Второй путь (безэталонный) - установление предела содержания примесей по отсутствию положительной реакции. При этом используют химические реакции, чувствительность которых ниже, чем предел обнаружения допустимых примесей.
Также используется хроматография (ФЭЖХ); оптические методы.
7. Приемы установления пределов допустимых примесей (эталонный и безэталонный).
Безэталонный метод – установление содержание примеси по отсутствию положительной химической реакции. В ФС указано, что данной примеси в препарате быть не должно.
Эталонный метод – основан на сравнении с эталоном, содержащим определенное количество открытых примесей. При использовании этого метода выполнение реакций осуществляется в одинаковых условиях, наблюдают окраску или помутнение, возникающее при добавлении соответствующего реактива. Определение примесей и приблизительную оценку их количсетва осуществляют колориметрическим или нефелометрическим методом.
8. Понятие «эталонный раствор», приготовление эталонных растворов, испытание на примеси хлоридов и сульфатов.
Эталонный раствор (раствор сравнения) – раствор, содержащий определенное количество открываемой примеси.
Изготовление эталонных растворов – содержание примеси и способ изготовления раствора указан в ГФ (ФС, ФСП).
Испытание на хлориды, сульфаты.
9. Понятие «эталонный раствор», приготовление эталонных растворов, испытание на примеси солей кальция, аммония.
Эталонный раствор (раствор сравнения) – раствор, содержащий определенное количество открываемой примеси.
Изготовление эталонных растворов – содержание примеси и способ изготовления раствора указан в ГФ (ФС, ФСП).
Испытание на соли аммония (реактив Несслера (K2[HgI4] + KOH) – желто-бурый осадок ([I2HgNH2]I)), соли кальция.
10. Понятие эталонный раствор, приготовление эталонных растворов, определение на примеси солей железа, цинка и тяжелых металлов.
Эталонный раствор (раствор сравнения) – раствор, содержащий определенное количество открываемой примеси.
Изготовление эталонных растворов – содержание примеси и способ изготовления раствора указан в ГФ (ФС, ФСП).
Испытание на соли железа (сульфаниловая кислота (NH2-Ar-SO3H)- комплексы, окраска зависит pH), соли цинка, тяжелые металлы.
11. Понятие растворимость в фармацевтическом анализе, ее использование в оценке качества ЛС.
Растворимость – свойство газов, жидкостей твердых веществ переходить в растворенное состояние. В ФА используется растворимость ЛВ в воде, 95% спирте, в органических растворителях (хлороформ, ацетон, эфир). Растворимость это не константа, а свойство, которое может служить ориентировочной характеристикой испытуемого вещества и несет дополнительную информацию о веществе. Термины ГФ XI: количество растворителя (мл), необходимое для растворения 1 г ЛВ; легко растворимые (до 1 мл), легко (1-10 мл), растворимые (10-30 мл), умеренно (30-100 мл), мало (100-1000 мл), очень мало (1000-10000 мл), нерастворимые (более 10000 мл).
Растворимость при постоянной температуре – 1 из основных характеристик доброкачественности ЛВ. Методика определения: навеску растворяют в V растворителя; если вещество не растворяется то увеличивают V растворителя. Растворимое ЛВ – отсутствие частиц при наблюдении в проходящем свете. Отклонение от правила: образование мути, растворение более 10 мин. (медленно растворимые).
Появление примесей изменяет растворимость.
12. Критерии оценки качества ЛС (прозрачность, степень мутности, окраска растворов).
Прозрачность, степень мутности определяют путем сравнения с испытуемой жидкости с растворителем, эталоном.
Испытание при освещении лампой матового стекла на черном фоне при вертикальном расположении пробирок. Прозрачная жидкость – нет нерастворенных частиц. Сравнение с растворителем для приготовления жидкости.
Окраска – визуально путем сравнения с эталонами (в равных количествах). Сравнение в пробирках одинакового d. При дневном отраженном свете на матово-белом фоне. Окраска должна быть идентична, не превышать интенсивность эталона.
Бесцветные жидкости – сверху на матово-белом фоне. Растворы – от соответствующего растворителя.
13. Определение летучих веществ и воды как критерии оценки качества лекарственных средств.
Физические методы (высушивание); химические – акваметрия (метод Фишера).
Высушивание – установление разности массы ЛВ до и после высушивания.
Метод Фишера – гигроскопичную и кристаллизационную воду определяют реактивом (SO2, I2, пиридин в метаноле).

Недостаток метода: герметичность, невозможность использования в присутствии веществ, реагирующих с компонентами реактива.
14. Зола, ее виды и способы определения при оценке качества лекарственных средств.
Зола – несгораемый минеральный остаток после сжигания вещества.
Зола: общая и сульфатная.
Общая – прокаливание навески ЛВ в тигле до постоянной массы, охлаждение в эксикаторе, взвешивание, добавление HCl, нагревание на водяной бане, фильтрование, сжигание осадка, взвешивание, определение содержания золы, нерастворимой в HCl.
Сульфатная – нагревание и прокаливание навески ЛВ, смоченной H2SO4 конц., нагревание на песчаной бане, прокаливание, охлаждение, установление массы. ФС – определение в сульфатной золе тяжелых Ме.
15. Определение содержания действующего вещества компонента методом аргентометрии на примере одного из лекарственных средств (химизм, способ фиксации точки эквивалентности).
Аргентометрия – основана на осаждении раствором AgNO3.
Hal– + AgNO3 AgHal + NO3–
Прямой метод (Мора) – нейтральная и щелочная среда; индикатор хромат калия; определение хлоридов, бромидов.
т.э. CrO42- + 2Ag+ = Ag2CrO4 (кирпичный)
Обратный метод (Фольгарда) – кислая среда; индикатор – железоаммонийные квасцы (NH4Fe(SO4)2*12H2O).
AgNO3ост. + KSCN = AgSCN + KNO3
т.э. (индикатор) Fe3+ + 3SCN- = Fe(SCN)3
ЛВ: NaCl, NaBr, KCl, KBr.
16. Определение содержания действующего компонента методом броматометрии на примере одного из ЛС.
Броматометрия (бромид-броматометрия) основана на использовании окислительных свойств или реакции замещения за счет образующегося свободного брома:
KBrO3 + 5KBr + 6HCl 3Br2 + 6KCl + 3H2O
Индикаторами при прямом титровании служат азокрасители, которые обесцвечиваются бромом в эквивалентной точке (метиловый красный). В случае обратного титрования эквивалентную точку устанавливают иодометрически по избытку титранта (бромата калия).
Количественное определение кислоты салициловой и натрия салицилата можно выполнить также бромид-броматометрическим методом, так как они являются
производными фенола:

Избыток брома затем определяют иодометрическим методом:
Br2 + 2KI = I2 + 2KBr
I2 + 2Na2S2O3 = 2NaI + Na2S4O6
17. Определение содержания действующего компонента методом йодхлорометрии на примере одного из ЛС.
Йодхлорометрия — отличается от йодометрии использованием в качестве титранта не раствора йода, а более устойчивого раствора йодмонохлорида. Аналогично йоду йодмонохлорид образует йодпроизводные органических оснований. Избыток титранта устанавливают йодометрически:

Фенол – избыток йода титруют раствором тиосульфата натрия.

18. Определение содержания действующего компонента методом комплексонометрии на примере одного из ЛС.
Метод основан на образовании прочных, растворимых в воде комплексов катионов металлов с трилоном Б — динатриевой солью этилендиаминтетрауксусной кислоты или другими комплексонами. Независимо от заряда катиона взаимодействие его с титрантом происходит в стехиометрическом соотношении 1:1.
Количественное определение висмута нитрата основного выполняют комплексонометрическим методом.

Выделяющаяся азотная кислота не мешает титрованию, так как соли висмута количественно взаимодействуют с ЭДТА*Na2 при pH 2–4. Вот почему в данном случае не требуется добавления буферного раствора. В эквивалентной точке выделяется свободный индикатор, который придает раствору желтую окраску.
Различают прямое; обратное (избыток ЭДТА оттитровывают MgSO4; индикатор магнезон); косвенное (по заместителю) – комплексы с Mg.
19. Цериметрия.
Цериметрия основана на использовании окислительных свойств титранта — соли церия (IV), который в кислой среде восстанавливается до церия (III):
Ce4+ + e = Ce3+
Индикатор - дифениламин, о-фенантролин, а при обратном титровании избыток титранта устанавливают иодометрически:
2Ce(SO4)2 + 2KI I2 + Ce2(SO4)3 + K2SO4
Резорцин
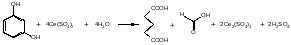
Избыток титранта устанавливают иодометрическим методом:
2Ce(SO4)2 + 2KI = I2 + Ce2(SO4)3 + K2SO4
I2 + 2Na2S2O3 = 2NaI + Na2S4O6
20. Йодометрия.
Йодометрия — метод, основанный на окислительных свойствах йода и восстановительных свойствах йодид-ионов:
I2 + 2e– 2I–
2I– – 2e– I2
Титрант — раствор йода (индикатор — крахмал) используют для прямого титрования неорганических и органических веществ, способных окисляться или образовывать с йодом продукты присоединения или замещения. Используют также обратное йодометрическое титрование. При этом избыток йода титруют раствором тиосульфата натрия:
I2 + 2Na2S2O3 2NaI + Na2S4O6
Восстановительные свойства йодида калия используют для количественного определения веществ, обладающих окислительными свойствами. Выделившееся эквивалентное количество йода оттитровывают тиосульфатом натрия.
Фенол – избыток йода титруют раствором тиосульфата натрия.

21. Йодатометрия.
Йодатометрия основана на окислении органических соединений йодатом калия. Избыток титранта устанавливают йодометрически:
KIO3 + 5KI + 6HCl 3I2 + 6KCl + 3H2O
Каптоприл

Титруют 0,1 М раствором йодата калия до появления голубой окраски, не исчезающей в течение 30 сек. Определение основано на окислении сульфгидрильной группы йодом:
KIO3 + 5KI + 3H2SO4 3I2 + 3K2SO4 + 3H2O
2R-SH + I2 R-S-S-R + 2HI
22. Определение содержания действующего компонента методом перманганатометрии на примере одного из ЛС.
Перманганатометрия основана на использовании окислительных свойств титранта — перманганата калия в кислой среде:
MnO4– + 8H+ + 5e = Mn2+ + 4H2O
Индикатором при прямом титровании служит сам титрант (появляется фиолетовое окрашивание), а при обратном титровании избыток титранта устанавливают иодометрическим методом.
Перманганатометрическое титрование применяется для определения очень сильных восстановителей, реагирующих с катионами железа(3+), переводя их в катионы железа(2+), которые оттитровывают перманганатом калия. Метод используется также для косвенных определений, например в феррометрии, суть которого заключается в восстановлении окислителей катионами железа(2+), избыток которого реагирует с перманганатом калия.
Обратным перманганатометрическим титрованием определяют восстановители, медленно реагирующие с KMnO4 – такие как иодиды, цианиды, тиоцианаты и т.п.
Количественное определение водорода перекиси — восстановителя выполняют прямым перманганатометрическим титрованием в кислой среде (до слабо-розового окрашивания):
2KMnO4 + 5H2O2 + 3H2SO4 = 2MnSO4 + K2SO4 + 8H2O + 5O2
23. Определение содержания действующего компонента методом нитритометрии на примере одного ЛС с первичной ароматической аминогруппой.
Метод количественного определения первичных и вторичных ароматических аминов, основанный на использовании титранта — раствора нитрита натрия, в присутствии нитрозил бромида:
NaNO2 + 0=N–Br + 2HCl О=N–Br + 2 NaCl + H2O
В этих условиях с ПЕРВИЧНЫМИ ароматическими аминами образуются диазосоединения (в кислой среде):
Ar–NH2 + 0=N–Br Ar–NH–N=O + HBr + HCl [Ar–N+N]Cl– + H2O
Эквивалентную точку устанавливают различными путями: потенциометрически, с помощью указанных в ФС внутренних индикаторов (тропеолин 00, нейтральный красный), с внешним индикатором (иодкрахмальная бумага). Титрование с иодкрахмальной бумагой ведут до тех пор, пока капля титруемого раствора, взятая через 1 мин после прибавления титранта, не вызовет тотчас же посинение бумаги:
2KI + 2NaNO2 + 4HCl I2 + 2NO + 2NaCl + 2KCl + 2H2O
При использовании внутренних индикаторов наблюдают изменение их окраски в эквивалентной точке.
Для количественного определения сложных эфиров n-аминобензойной кислоты ФС рекомендует нитритометрический метод. При определении бензокаина, прокаина гидрохлорида, как и других первичных ароматических аминов, происходит образование солей диазония:
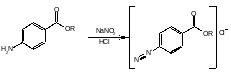
24. Определение содержания действующего компонента методом нитритометрии на примере одного ЛС со вторичной ароматической аминогруппой.
Метод количественного определения первичных и вторичных ароматических аминов, основанный на использовании титранта — раствора нитрита натрия, в присутствии нитрозил бромида:
NaNO2 + 0=N–Br + 2HCl О=N–Br + 2 NaCl + H2O
ВТОРИЧНЫЕ ароматические амины в этих условиях образуют N-нитрозосоединения:
Ar–NH(R) + О=N–Br Ar–N(R)NO + HBr
Эквивалентную точку устанавливают различными путями: потенциометрически, с помощью указанных в ФС внутренних индикаторов (тропеолин 00, нейтральный красный), с внешним индикатором (иодкрахмальная бумага). Титрование с иодкрахмальной бумагой ведут до тех пор, пока капля титруемого раствора, взятая через 1 мин после прибавления титранта, не вызовет тотчас же посинение бумаги:
2KI + 2NaNO2 + 4HCl I2 + 2NO + 2NaCl + 2KCl + 2H2O
При использовании внутренних индикаторов наблюдают изменение их окраски в эквивалентной точке.
Для количественного определения сложных эфиров n-аминобензойной кислоты, ФС рекомендует нитритометрический метод. Тетракаин, как и другие вторичные амины, образует N-нитрозосоединение:

Точку эквивалентности при титровании тетракаина гидрохлорида устанавливают с помощью внешних индикаторов.
25. Метод кислотно-основного титрования в неводных средах на примере слабой органической кислоты.
Титрование в среде неводных растворителей (неводное титрование)
Метод позволяет количественно определить органические вещества, проявляющие в водной среде очень слабые основные или кислотные свойства. В качестве титрантов используют растворы сильных кислот или сильных оснований.
Неводное титрование органических веществ, проявляющих кислотные свойства (фенолы, барбитураты, карбоновые кислоты, сульфаниламиды и др.) выполняют, используя в качестве растворителя диметилформамид или его смесь с бензолом. Титрантом служит раствор гидроксида натрия в смеси метанола и бензола или раствор метилата натрия. В качестве индикатора используют тимоловый синий.
R–OH + O=CН–N(CH3)2 R–O– + O=CН–NH+(CH3)2
R–O– + CH3ONa R–ONa + CH3O–
CH3O– + O=CН — NH+(CH3)2 CH3OH + O=CН–N(CH3)2
Суммарно процесс нейтрализации фенолов (енолов) можно представить так:
R–OH + CH3ONa R–ONa + CH3OH
26. Метод кислотно-основного титрования в неводных средах на примере слабого органического основания.
Неводное титрование органических оснований (R3N) и их солей (R3N · HA) выполняют, используя в качестве растворителей безводные уксусную кислоту, уксусный ангидрид, муравьиную кислоту или их сочетания. Титрантом служит раствор хлорной кислоты, индикаторами — кристаллический фиолетовый, тропеолин, метиловый оранжевый.
Титрование слабых органических оснований хлорной кислотой в среде ледяной уксусной кислоты включает несколько этапов
1. Растворение HClO4 в ледяной CH3COOH:
HClO4 + CH3COOH ⇄ CH3COOH2+ + ClO4–
2. Растворение основания (R3N) в ледяной CH3COOH:
R3N + CH3COOH [R3NH]+ + CH3COO–
3. Взаимодействие ацетоний- и ацетат-ионов:
CH3COOH2+ + CH3COO– 2CH3COOH
4. Взаимодействие протонированного амина с хлорат-ионом:
[R3NH]+ + ClO4– [R3NH]+ · ClO4–
—————————————————————————————
R3N + HClO4 [R3NH]+ · ClO4–
Очень слабые органические основания (рК>12) необходимо титровать хлорной кислотой в среде уксусного ангидрида (УА), т.к. он более активно (чем ледяная уксусная кислота) усиливает основные свойства аминов.
1. Взаимодействие HClO4 с УА:
HClO4 + (CH3CO)2O ⇄ (CH3CO)2OH+ + ClO4–
2. Растворение амина (R3N) в УА:
R3N + (CH3CO)2O ⇄ R3NCH3CO+ + CH3COO–
ацетилий-ион
3. Взаимодействие кислоты с основанием:
(CH3CO)2OH+ + CH3COO– ⇄ (CH3CO)2O + CH3COOH
4. Взаимодействие ацетилий-иона с хлорат-ионом:
R3NCH3CO+ + ClO4– + CH3COOH [R3NH]+ · ClO4– + (CH3CO)2O
—————————————————————————————
R3N + HClO4 [R3NH]+ · ClO4–
Соли органических оснований с галогеноводородными кислотами (R3N · HX) проявляют кислотные свойства даже в неводной среде. Поэтому их титруют в присутствии ацетата ртути (II), который нейтрализует галогенпроизводную кислоту. Образующийся ацетат органического основания оттитровывают хлорной кислотой:
2R3N · HX + (CH3COO)2Hg HgX2 + 2[R3NH]+ · CH3COO–
2[R3NH]+ · CH3COO — + 2HClO4 2[R3NH]+ · ClO4– + 2CH3COOH
Неводное титрование галогеноводородов может быть выполнено без добавления ацетата ртути, если в качестве протогенных растворителей использовать безводную муравьиную кислоту в присутствии уксусного ангидрида.
27. Биологические методы анализа (определение активности антибиотиков, сердечных гликозидов, витаминов).
Антибиотиками называют вещества, продуцируемые микроорганизмами, высшими растениями, животными тканями в процессе их жизнедеятельности и обладающие способностью оказывать на микроорганизмы, простейшие, некоторые вирусы избирательное бактериостатическое или бактерицидное действие. Способность антибиотиков проявлять бактериостатическое или бактерицидное действие в отношении болезнетворных микроорганизмов, не оказывая при этом токсического действия на организм человека, используют для лечения различных заболеваний.
Методы анализа антибиотиков
Активность устанавливают диффузионным или турбидиметрическим методами. ГФ XI рекомендует для количественного определения метод диффузии в агар, заключающийся в сравнении действия определенных концентраций испытуемого и стандартного образца антибиотика на тест-микроорганизм.
Поскольку состав агаровой среды и условия выполнения биологического испытания одинаковы, величина зоны диффузии (в которой развитие тест-микроорганизма подавляется антибиотиком) зависит только от химической природы антибиотика и его концентрации.
Единица действия (ЕД) представляет собой меру, которой выражается биологическая активность антибиотиков. За ЕД принимают минимальное количество антибиотика, подавляющего развитие тест-микроорганизма в определенном объеме питательной среды.
К ускоренным микробиологическим методам относят методы, основанные на подавлении изменений рН питательной среды в процессе роста тест-микроорганизмов (уреазный метод).
Сердечные гликозиды - безазотистые соединения растительного происхождения, характеризующиеся кардиотоническим действием. Данные препараты играют исключительно важную роль в терапии больных с острой и хронической сердечной недостаточностью любого генеза. При определении активности лекарственного сырья и многих препаратов сердечных гликозидов используют биологическую стандартизацию. Наиболее часто активность сердечных гликозидов выражают в лягушачьих единицах действия (ЛЕД) и кошачьих единицах действия (КЕД). Одна ЛЕД соответствует минимальной дозе стандартного препарата, в которой он вызывает остановку сердца у большинства подопытных лягушек, кошек, голубей. Так, размельченный порошок листьев наперстянки по активности соответствует такой пропорции: один грамм порошка листьев равен 50-66 ЛЕД или 10-13 КЕД. В процессе хранения активность листьев уменьшается.
Витамины представляют собой группу веществ различной химической структуры, необходимых в малых количествах для нормальной жизнедеятельности организма. Ряд витаминов входят в состав ферментных систем и являются своеобразными биологическими катализаторами химических или фотохимических процессов, происходящих в живой клетке (тиамин, рибофлавин, пиридоксин, пантотеновая кислота и др.).
Для качественной и количественной оценки витаминов в природных источниках используют как биологические, так и физико-химические методы. Принцип оценки биологической активности заключается в том, что животных (крыс, голубей, морских свинок) переводят на диету, содержащую белки, жиры, углеводы, минеральные соли и все витамины, кроме исследуемого. Затем устанавливают, какое количество испытуемого витамина может излечить или предохранить животное от авитаминоза. Параллельно проводят аналогичное испытание со стандартным препаратом.
Биологический метод оценки активности витаминов очень трудоемок, точность его сравнительно невелика. Поэтому для испытания подлинности и количественного определения витаминов обычно используют физические, химические и физико-химические методы.
28. Стабильность и сроки годности ЛС (влияние влаги, CO2, света, кислорода воздуха, примесей).
Под сроком годности лекарственных средств понимают период времени, в течение которого они должны полностью сохранять свою терапевтическую активность, безвредность и по уровню качественных и количественных характеристик соответствовать требованиям ГФ или ФС (ФСП), в соответствии с которыми были выпущены и хранились в условиях, предусмотренных указанными статьями.
По истечении срока годности ЛС не может быть использовано без переконтроля качества и соответствующего изменения установленного срока годности. Существует определенная взаимосвязь между понятием «срок годности», имеющим временной смысл, и понятием «стабильность», обусловливающим качество ЛС (его устойчивость).
Температура – с увеличением увеличивается скорость реакции; с понижением (понижается активность MgSO4, CaCl2, раствора адреналина).
Свет – повышается скорость разложения; кристаллические сухие вещества более устойчивы, чем растворы; изменение цвета при длительном освещении; некоторые вещества сохраняют свою активность (содержащие железо, при этом повышается их стабильность).
Влага – снижает фармакологическую активность; + и – влияет на ЛВ; гигроскопичность.
Окисление — процесс, являющийся одной из причин разложения ЛВ. Некоторые из них (производные фенолов) окисляются, находясь в кристаллическом состоянии. Процесс окисления заметно активизируется при растворении. Особенно легко окисляются ЛВ, проявляющие активные восстановительные свойства (альдегиды, гидразиды, производные фенотиазина и др.).
Система мер, направленных на предохранение ЛВ от окисления, сводится прежде всего к уменьшению влияния атмосферного кислорода или максимальному удалению примесей, катализирующих процесс окисления. Используя окислители, можно смоделировать процесс окисления. Если затем сравнить полученные продукты окисления стандартного образца и продукты разложения ЛВ, то можно сделать заключение о механизме процесса окисления. Это позволяет решать вопрос о путях стабилизации, так как станут известны факторы, влияющие на скорость реакции окисления.
Методы повышения стабильности:
1) физические (твердые вещества – в плотно укупоренной таре; суспензии – в сухом состоянии; инъекции – в ампулах запечатанных);
2) химические (окисление, металлы).
29. Фармакокинетика и биодоступность.
Фармакокинетика – раздел фармакологии о всасывании, распределении, депонировании, метаболизме и выделении ЛВ.
Проведение фармакокинетических исследований возможно только на основе применения современных методов биофармацевтического анализа, позволяющих проследить процесс всасывания и распределения ЛВ в органах и тканях. Они включают выяснение влияния различных биофармацевтических факторов на терапевтическую эффективность ЛВ; изучение их биологической доступности и разработку методов ее определения; создание способов определения ЛВ и их метаболитов в биологических жидкостях.
На фармакокинетику ЛВ оказывают влияние различные факторы: возрастные, генетические, половые, масса тела, питание, беременность, а также различные патологические процессы, например заболевания печени, почек, сердечно-сосудистой системы, желудочно-кишечного тракта, эндокринные, инфекционные и другие заболевания.
Биодоступность – количество неизменного вещества, которое достигло плазмы крови, относительно исходной дозы препарата.
Одним из основных этапов любого исследования биологической доступности ЛС является использование биофармацевтического анализа для определения концентрации ЛВ (метаболита) в биологических жидкостях.
30. Рефрактометрия
Рефрактометрия основана на наличии зависимости величины показателя преломления света от концентрации раствора испытуемого вещества. Показатель преломления зависит также от температуры, длины волны света, концентрации вещества и природы растворителя. Рефрактометрию используют для установления подлинности лекарственных веществ по молярной рефракции. Для количественного определения выбирают интервал линейной зависимости между концентрацией раствора и коэффициентом преломления. В этом интервале концентрацию (х) вычисляют по формуле: х=(n – nO)/F, где n — показатель преломления раствора вещества; nO — показатель преломления растворителя; F — фактор, равный величине прироста показателя преломления при увеличении концентрации вещества на 1% (устанавливается экспериментально).
Рефрактометрические определения выполняют на рефрактометрах, при стабильной температуре (200,3ОC) и длине волны линии D спектра натрия (589,3 нм) в диапозоне показателей преломления от 1,3 до 1,7. Прибор юстируют по эталонным жидкостям или воде очищенной, для которой nD20 = 1,3330.
31. Спектрофотометрия в уф-, видимой, ик-областях спектра в оценке качества лс.
Используют спектрофотометрические методы анализа по поглощению веществами монохроматического электромагнитного излучения.
Фотометрические методы анализа основаны на использовании закона Бугера-Ламберта-Бера:

В случае несоответствия закону вначале с помощью стандартного раствора устанавливают зависимость оптической плотности от концентрации, а затем строят калибровочный график, с помощью которого выполняют расчеты.
Диапазоны света:

Спектрофотометрия в УФ- и видимой областях – 1 из широко используемых физико-химических методов в фармацевтическом анализе.
Анализируемые ЛВ должны иметь в структуре молекулы хромофорные группы (сопряженные связи, ароматическое ядро и др.), обусловливающие различные электронные переходы в молекулах и поглощение электромагнитного излучения.
Кривая зависимости интенсивности светопоглощения от длины волны (нм) называется спектром поглощения вещества и является его специфической характеристикой. Измерение спектров поглощения растворов анализируемых веществ в УФ (190-380 нм) и видимой (380-780 нм) областях производят с помощью спектрофотометров различных марок (СФ-26, СФ-46 и др.). В качестве растворителей используют свободные от примесей воду, растворы кислот и щелочей, этанол, хлороформ и другие органические растворители.

Удельный показатель поглощения представляет собой величину оптической плотности раствора, содержащего 1,0 г вещества в 100 мл раствора, измеренную в кювете с рабочей длиной 1 см. Установив по стандартному образцу величину E и преобразовав эту формулу, можно рассчитать концентрацию анализируемого вещества с относительной погрешностью до ±2%.
Константа измеряется в различных единицах; в молях – молярный коэффициент поглощения, в % - удельный показатель поглощения
Идентификацию ЛВ можно провести по, Е, характеру спектральных кривых в различных растворителях, положению максимума и минимума светопоглощения или их отношению (при различных длинах волн). Для количественного спектрофотометрического анализа важен выбор аналитической полосы поглощения. Последняя должна быть свободна от наложения полос поглощения других компонентов смеси и иметь достаточно высокий удельный показатель поглощения анализируемого вещества.
Спектрофотометрия в ИК-области. Природа полос поглощения в ИК области связана с колебательными переходами и изменением колебательных состояний ядер, входящих в молекулу поглощающего вещества. Поэтому поглощением в ИК-области обладают молекулы, дипольные моменты которых изменяются при возбуждении колебательных движений ядер. Область применения ИК-спектроскопии аналогична, но более широка, чем у УФ-метода. ИК-спектр однозначно характеризует всю структуру молекулы, включая незначительные ее изменения. Важные преимущества ИК-спектроскопии — высокая специфичность, объективность полученных результатов, возможность анализа веществ в кристаллическом состоянии. Для измерения ИК-спектров на однолучевых или двулучевых ИК-спектрофотометрах используют взвеси веществ в вазелиновом масле или помещают анализируемое вещество между пластинами из бромида калия.
Каждый ИК-спектр представляет собой серию полос поглощения, максимумы которых определяются волновым числом, измеряемым в см-1, и определенной интенсивностью. Для анализа ЛB обычно используют спектральную область от 4000 до 400 см-1.
ГФ XI рекомендует два способа установления подлинности по ИК-спектрам. Один из них основан на сравнении зарегистрированных в идентичных условиях ИК-спектров испытуемого ЛB и его стандартного образца. Второй способ заключается в сравнении ИК-спектра испытуемого ЛB с его стандартным спектром, прилагаемым к ФС и зарегистрированным в соответствии с указанными в ней требованиями.
УФ |
Видимый |
ИК |
|
p-электроны |
Функциональные группы |
||
Определение подлинности и количественное определение |
Определение подлинности |
||
Истинные растворы |
Таблетки |
||