

Лекция 1
1. Кинетические закономерности электрохимического растворения
и осаждения металлов
1.1. Введение
Процессы осаждения металлов на твёрдых металлических электродах, так же как и процессы анодного растворения металлов и сплавов, являются одними из наиболее сложных электрохимических реакций. Они, как правило, протекают через несколько стадий. Эти стадии включают процессы диффузии, адсорбции1, химической реакции, разряда-ионизации и кристаллизации, участвующих в электрохимическом процессе частиц. Соотношение скоростей этих стадий определяет кинетику процесса, как катодного осаждения, так и анодного растворения металла.
Электроосаждение металлов из водных растворов обычно сопровождается протеканием параллельной реакции выделения водорода или участием в реакции других частиц, находящихся в электролите. В результате протекания реакции происходят изменение состава раствора у поверхности электрода и изменения состояния поверхности. Это особенно сильно проявляется в первые моменты электролиза после включения тока. Все предшествующие электрокристаллизации металла стадии влияют на неё. Таким образом они определяют структуру, физико-механические и химические свойства электроосаждённого металла.
Знание кинетических закономерностей электроосаждения и растворения металлов позволяет выбирать рациональную скорость проведения процесса, характеризующегося плотностью тока. А также установить другие параметры проведения процесса – концентрации компонентов электролита, температуры электроосаждения и т.д. – с целью получения осадков или поверхности с заданными свойствами.
1.2. Равновесие на границе «металлический электрод –
раствор электролита»
При погружении металла в раствор электролита на нём, выступающем как электрод, устанавливается равновесный или стационарный потенциал. Его установление связано с обменом зарядами между металлом и его ионами в растворе. Для установления равновесного потенциала необходимо выполнение двух условий:
первое, наличие только одной реакции обмена зарядами между металлическим электродом и раствором;
второе, достаточно высокая скорость этого обмена.
Время установления потенциала зависит от скорости обмена. Если на электроде протекает только одна обменная реакция типа:
Mn+ + ne = M, (*)
то выражение для равновесного электродного потенциала (уравнение Нернста) имеет вид:
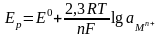 ,
,
где
Ер
– равновесный
электродный потенциал, измеренный
относительно нормального водородного
электрода сравнения; Е0
– стандартный
электродный потенциал; R
= 8,314 Дж/(Кмоль)
– универсальная газовая постоянная; Т
– абсолютная температура; п
– число электронов, принимающих участие
в электродной реакции; F
= 96493 К/моль – число Фарадея;
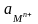 -
активность ионов металла в растворе;
2,3 – переводной множитель из натуральных
логарифмов в десятичные.
-
активность ионов металла в растворе;
2,3 – переводной множитель из натуральных
логарифмов в десятичные.
Для более сложных электродных реакций, когда в них участвуют комплексные ионы металла, например типа
MLi + ne- = M + iL,
уравнение Нернста имеет следующий вид:
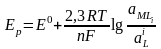 ,
где
,
где
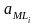 ,
,
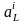 - активности соответственно комплексных
ионов металла и лиганда.
- активности соответственно комплексных
ионов металла и лиганда.
(Лиганды – молекулы или ионы, связанные с центральным ионом-комплексообразователем в комплексном соединении).
Равновесный потенциал устойчив во времени, не зависит от перемешивания раствора и возвращается к исходному значению после отключения тока через электрохимическую систему.
Электроосаждение металлов проводят из растворов, содержащих как простые, так и комплексные ионы металлов. На присутствие в растворе комплексных ионов указывает сдвиг равновесного потенциала в отрицательную сторону по сравнению с раствором, содержащим простые ионы.
Ионы большинства металлов вступают в растворе в реакцию комплексообразования с различными ионами и молекулами. В качестве лигандов могут выступать молекулы растворителя (воды), ионы гидроксония и гидроксила, анионы и катионы, молекулы и ионы органических соединений. Реакция образования комплексного иона металла с лигандами записывается в виде:
Mn+ + iL = MLi
При комплексообразовании в растворе устанавливается равновесие между всеми присутствующими частицами. Равновесие характеризуется общей концентрационной константой устойчивости (образования) комплекса:
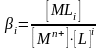 .
.
В квадратных скобках записаны равновесные концентрации частиц, участвующих в реакции комплексообразования.
Величина равновесного потенциала при комплексообразовании определяется выражением:
 ,
,
где:
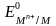 – стандартный потенциал реакции (*);
– стандартный потенциал реакции (*);
i – константа устойчивости i – го комплекса;
 – аналитическая
концентрация ионов металла.
– аналитическая
концентрация ионов металла.
Поскольку
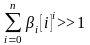 ,
то равновесный потенциал в растворе,
содержащем комплексные ионы, всегда
отрицательнее, чем в растворе простых
ионов. Поэтому сдвиг потенциала в
отрицательную сторону при увеличении
концентрации какого-либо компонента
раствора однозначно указывает на
протекание реакции комплексообразования.
,
то равновесный потенциал в растворе,
содержащем комплексные ионы, всегда
отрицательнее, чем в растворе простых
ионов. Поэтому сдвиг потенциала в
отрицательную сторону при увеличении
концентрации какого-либо компонента
раствора однозначно указывает на
протекание реакции комплексообразования.
1.3. Квазиравновесие на границе «электрод-раствор».
Природа бестокового потенциала
Как правило, в растворах для электрохимического осаждения металлов, а также для электрохимической и химической обработки металлов на электродах в отсутствие тока устанавливается не равновесный, а станционарный потенциал.
Стационарный потенциал, в отличие от равновесного, характеризуется протеканием на одном и том же электроде двух и более реакций, которые называются сопряженными. Одной из реакций является разряд-ионизация металла (*):
Mn+ + ne = M.
Другими реакциями могут быть реакции самого различного типа. Например, в кислых водных растворах – катодная реакция разряда ионов гидроксония и ионизации водорода.
Величина стационарного потенциала определяет не только тип сопряжённой реакции, но и скорость растворения металла, то есть скорость его коррозии. Поэтому стационарный потенциал часто называют коррозионным. Так как при этом протекает как минимум две реакции, и потенциал приобретает значение, расположенное между значениями равновесных потенциалов этих реакций, то его можно назвать компромиссным. Если неизвестна природа реакций, приводящих к установлению потенциала, его можно назвать бестоковым.
Общий состав раствора определяет значение стационарного потенциала и тип обменных реакций. Допустим, что в растворе присутствуют компоненты, способные восстанавливаться на поверхности металла, то есть такие, равновесный потенциал которых положительнее равновесного потенциала металла.2 В этом случае на электроде будет протекать реакция восстановления этих компонентов и сопряжённая реакция растворения металла.
Процессы химического травления металлов протекают с сопряжённой реакцией восстановления какого-либо окислителя. Для электроотрицательных металлов такой реакцией обычно является выделение водорода. Для электроположительных – окислительно-восстановительная реакция, равновесный потенциал которой положительнее равновесного потенциала металла. Например, при растворении меди в растворах хлорида железа сопряжённой реакцией является восстановление ионов Fe+3 до ионов Fe+2.
1.4. Измерение Э.Д.С. и электродных потенциалов
Измерять прямым методом потенциал электрода невозможно. непосредственно можно измерить лишь Э.Д.С. элемента. Непременным условием измерения Э.Д.С. является отсутствие тока в момент измерения в цепи элемента. В этом случае Э.Д.С. равна разности равновесных потенциалов двух электродов, из которых составлена цепь. При измерении потенциалов отдельных электродов составляют гальванический элемент из испытуемого электрода и электрода сравнения. В качестве электродов сравнения используют электроды второго рода.
Электрод первого рода – это система из металла, погружённого в раствор, содержащий ионы этого элемента. Например, на медном электроде в растворе соли меди устанавливается равновесие: Cu ↔ Cu2+ + 2e. К электродам первого рода относят, в частности, амальгамные.
Электроды второго рода – это системы из металла, покрытого слоем его трудно растворимой соли (или оксида) и погружённого в раствор, содержащий анионы этой соли (для оксида – ионы ОН–).
Потенциал электродов второго рода зависит от активной концентрации анионов в растворе. Наиболее широко применимы хлорсеребряный (Ag, AgCl/KCl) и каломельный (Hg, Hg2Cl2/KCl) электроды. В насыщенном растворе хлорида калия при 25С EAgCl = 0,2438 В, EHg2Cl2 = 0,200 В относительно стандартного водородного электрода, потенциал которого равен нулю.
По измеренной величине Э.Д.С. = Е и значению потенциала электрода сравнения ЕЭС рассчитывается неизвестный потенциал исследуемого электрода ЕИЭ:
ЕИЭ = Е + ЕЭС.
Измеряемая Э.Д.С. = Е будет иметь отрицательный знак при ЕЭС > ЕИЭ и положительный знак при ЕЭС < ЕИЭ.
ТОЭ Лекция 2-3
1.5. Двойной электрический слой.
1.5.1. Возникновение двойного электрического слоя (ДЭС).
Если
металлический электрод поместить в
раствор соли какого-либо металла, то
начинается переход атомов металла с
электрода в раствор и обратный переход
ионов металла из раствора на электрод.
Скорости перехода характеризуются
своим током:
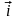 и
и
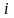 .
Эти скорости не равны. Их разность
называется током обмена
.
Эти скорости не равны. Их разность
называется током обмена
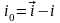 .
Через некоторое время скорости
уравниваются и электрод переходит в
равновесное состояние, приобретая
потенциал.
.
Через некоторое время скорости
уравниваются и электрод переходит в
равновесное состояние, приобретая
потенциал.
В состоянии равновесия возможны следующие случаи.
Первый случай. Положительно заряженные ионы металла частично перешли из раствора на электрод. Электрод заряжается положительно по отношению к раствору и притягивает к своей поверхности отрицательно заряженные ионы. У поверхности возникает обкладка из отрицательно заряженных ионов и скачок электрического потенциала.
Часть отрицательно заряженных ионов будет притянута к поверхности электрода на минимальное расстояние 0 , равное их радиусу. На расстоянии бóльшем, чем 0 присутствуют как анионы, так и катионы. На некотором расстоянии от электрода их концентрации сравняются, и раствор станет электронейтральным.
Заряженная поверхность электрода вместе с прилегающим к электроду противоположно заряженным слоем раствора называется двойным электрическим слоем (ДЭС).
Часть ДЭС, которая образуется непосредственно прилегающими к электроду ионами (на расстоянии 0 ) называется плотной частью ДЭС. В этой части потенциал между электродом и плоскостью, проходящей через центры прилегающих к нему ионов, изменяется линейно. Скачок потенциала в плотной части ДЭС обозначается . Часть ДЭС, расположенная за плотной частью, называется диффузной. Изменение потенциала с расстоянием от электрода в ней нелинейно и обозначается буквой 1. Полный скачок потенциала аб на границе электрода с раствором называется абсолютным скачком потенциала. Он равен аб = + 1.
Второй случай. Когда часть атомов металла перейдёт в виде положительных ионов в раствор. Электрод заряжается отрицательно по отношению к раствору. Притягивающиеся к поверхности катионы образуют положительно заряженную ионную обкладку электрода.
1.5.2. Строение ДЭС
При прохождении через границу раздела «электрод-электролит» постоянного электрического тока происходит изменение скачка потенциала «электрод-электролит». Оно сопровождается изменением строения ДЭС. Основная особенность ДЭС – наличие в нём избытка ионов того или иного знака. Вне ДЭС раствор электронейтрален.
Изменение концентрации катионов и анионов в ДЭС при отрицательно заряженном электроде показано на рисунке.
СК, Са – концентрации катионов и анионов;
тт – поверхность электрода;
пп - граница плотной части ДЭС;
пп – граница диффузной части ДЭС;
х – расстояние от электрода;
- толщина ДЭС;
0 – толщина плотной части ДЭС;
1 – толщина диффузной части ДЭС.
К отрицательно заряженному электроду притягиваются катионы. Поэтому их концентрация в слое раствора, прилегающем к электроду, больше, чем анионов. Следовательно, прилегающий к отрицательно заряженному электроду раствор имеет избыточный положительный заряд. По мере удаления от электрода концентрация катионов уменьшается, а анионов возрастает. На расстоянии «тп» концентрации катионов и анионов сравниваются, и раствор становится электронейтральным.
У положительно заряженного электрода ситуация аналогична, только катионы и анионы меняются местами.
Толщина диффузной части ДЭС зависит от концентрации раствора и уменьшается с увеличением концентрации. В очень разбавленных растворах толщина диффузной части (10-6 – 10-4) см. В сильно концентрированных растворах диффузная часть вырождена, толщина ДЭС приближается к толщине плотной части, а изменение потенциала в ДЭС приближается к скачку потенциала в плотной его части.
1.5.3. Ёмкость ДЭС
При достаточной концентрации раствора толщина ДЭС равняется толщине его плотной части, то есть радиусу прилегающих к электроду ионов. Такой ДЭС можно уподобить плоскому электрическому конденсатору. Одна его обкладка – заряженный электрод, другая – слой прилегающих к электроду ионов. Применим уравнение для плоского конденсатора:
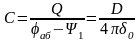 ,
[Ф/см2]
(**)
,
[Ф/см2]
(**)
Q – заряд 1 см2 поверхности электрода; С – ёмкость ДЭС у электрода с поверхностью 1 см2; D – диэлектрическая проницаемость среды между обкладками; 0 – расстояние между обкладками (толщина плотной части ДЭС).
Таким образом, ёмкость не должна зависеть от потенциала. Однако в плотной части молекулы и ионы находятся в электрическом поле большой напряжённости и сильно поляризуются. В результате величины D и 0 меняются и зависят от потенциала. Так, например, диэлектрическая проницаемость воды в плотной части составляет 3-10 против 81 в объёме раствора.
Так как анионы менее гидратированы, чем катионы, то их гидратная оболочка сильнее деформируется в электрическом поле. Поэтому анионы прочнее адсорбируются на металле и повышают ёмкость. Катионы, как правило, слабо адсорбируются и мало влияют на ёмкость.
В разбавленных растворах скачком потенциала в диффузной части ДЭС нельзя пренебречь. Здесь:
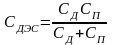 ,
где
,
где
СП – ёмкость плотной части, определяемой по (**); СД – ёмкость диффузной части ДЭС. То есть эти части рассматриваются как параллельно включённые конденсаторы.
Толщина диффузной части обычно не превышает 10-4 см.
Различают:
1. Среднюю ёмкость электрода
 ,
,
где Q – изменение заряда поверхности электрода; - изменение потенциала электрода.
2. Интегральную ёмкость
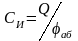 ,
,
где Q – заряд электрода; аб – значение абсолютного потенциала электрода.
3. Дифференциальную ёмкость
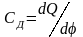 ,
,
то есть ёмкость ДЭС, соответствующую данному потенциалу электрода. Здесь dQ и d - бесконечно малые изменения количества электричества и потенциала электрода за бесконечно малое время поляризации электрода.
Способы измерения ёмкости ДЭС достаточно сложны и требуют высокой квалификации исполнителя.
1.6. Основные закономерности восстановления ионов
и растворения металлов
Восстановление и окисление ионов металла происходит при некотором сдвиге потенциала электрода от равновесного или стационарного значения в отрицательную сторону. Сдвиг потенциала называется перенапряжением или поляризацией электрода:
= Е – Еравн,
= Е – Естацон,
где - перенапряжение (поляризация );
Е – потенциал электрода при пропускании тока.
Для катодных реакций поляризация и перенапряжение отрицательные величины, для анодных – положительныея.
Сдвиг потенциала от стационарного значения должен вызвать протекание внешнего тока (катодного или анодного). Значение этого тока зависит от многих факторов и определяется механизмом реакции. Механизм реакции, в свою очередь, определяется элементарными стадиями, через которые она протекает. Значение тока, отнесённое к единице поверхности электрода Sэлектрода (так называемая плотность тока), однозначно определяет скорость электрохимической реакции:
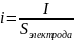 ,
[A/см2
или А/дм2]
,
[A/см2
или А/дм2]
Основными элементарными стадиями процесса восстановления ионов металла в простейшем случае являются:
доставка ионов из объёма раствора к поверхности металла;
разряд ионов;
образование кристалла.
В более сложных случаях, например при выделении металлов из комплексных ионов, разряду могут предшествовать гомогенные или гетерогенные химические реакции. Процесс разряда может сопровождаться также адсорбцией ионов металла или компонентов раствора на электроде и другими поверхностными явлениями.
Основными элементарными стадиями процесса анодного растворения в простейшем случае являются:
доставка катионов из объёма раствора к поверхности металла;
разряд-ионизация ионов;
отвод ионов металла в объём раствора.
Параллельно процессу ионизации металла может протекать реакция образования окисла на анодной поверхности.
Характер зависимости плотности тока (то есть скорости реакции восстановления) от потенциала электрода, концентрации компонентов раствора, гидродинамических условий, температуры, наличия ПАВ определяется природой наиболее медленной стадии, через которые протекает реакция.
В общем случае величина потенциала электрода складывается из величин перенапряжений элементарных стадий.
1.6.1. Перенапряжение диффузии
Пусть замедленной стадией процесса восстановления ионов металла является их доставка к поверхности электрода. Процесс доставки участвующих в реакции ионов или молекул к электроду осуществляется диффузией, миграцией и конвекцией.
Диффузия – перемещение частиц под действием градиента химического потенциала, или, приближённо, градиента концентрации, возникающего в растворе в результате его качественной или количественной неоднородности.
Миграция – передвижение ионов под действием градиента электрического поля, возникающего в электролите при прохождении тока через электрохимическую систему.
Конвекция – перенос частиц растворённого вещества вместе с потоком движущейся жидкости.
Перенапряжение диффузии имеет вид:
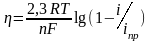 ,
,
где: i – плотность тока; iпр – предельная плотность тока диффузии (когда концентрация вступающих в реакцию частиц у поверхности электрода стремится к нулю).
Зависимость плотности тока (а) и логарифма плотности тока (б) от перенапряжения при концентрационной поляризации (замедленная диффузия).
Зависимость потенциала от плотности тока при замедленной диффузии имеет следующий вид:
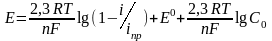 ,
,
где Е0 – стандартный потенциал реакции «металл - ионы металла»;
С0 – концентрация потенциалопределяющих ионов в объёме электролита.
Видно, что зависимость потенциала электрода от плотности тока такая же, как для перенапряжения. Однако при изменении концентрации электролита поляризационные кривые будут смещаться. Смещение эквивалентно изменению равновесного потенциала в зависимости от концентрации компонента. Также происходит пропорциональное изменение предельной плотности тока.
Влияние концентрации электролита
на зависимость плотности тока
от потенциала при замедленной
диффузии
C1>C2>C3
Повышение концентрации ионов металла в электролите вызывает сдвиг равновесного потенциала и возрастание предельного тока. Если после достижения предельного тока продолжать увеличивать ток, то потенциал сместится до значения, при котором будет протекать другая электродная реакция. На катоде чаще всего – выделение водорода. При этом происходит подщелачивание прикатодного слоя. В его результате при превышении в прикатодном слое рН гидратообразования, на поверхности электрода осаждается гидроксид металла. Он включается в металлический осадок. При сильном подщелачивании гидроксид экранирует поверхность. В результате ток падает. Это весьма негативное явление, так как осаждение металла в области предельного тока обычно сопровождается образованием рыхлых губчатых или дендридообразных осадков.
При сдвиге потенциала электрода от равновесного в анодную сторону ток теоретически должен непрерывно возрастать. Но здесь начинает проявляться замедленность стадии отвода ионов от электрода в объём электролита. Превышается растворимость соли металла, и она выпадает на поверхности анода. Наступает солевая пассивация.
В практике электроосаждения металлов никогда не реализуются условия, когда доставка вещества к электроду происходит только диффузией. Вклад миграции в скорость доставки ионов к поверхности катода достаточно велик. При восстановлении на катоде катионов металла диффузия и миграция направлены к электроду. В результате поток ионов возрастает. Это приводит как к увеличению тока при заданном потенциале, так и к увеличению предельного тока. При восстановлении на катоде комплексных ионов, их перенос диффузией направлен к катоду. Под действием же сил электрического поля они будут двигаться к аноду. Аддитивность действия этих двух процессов приводит к снижению потока вещества к поверхности электрода и уменьшению тока и предельного тока. Аналогичные явления протекают и при растворении анодов.
Поэтому введение в электролит «нейтральных» солей (например для повышения проводимости раствора), или увеличение концентрации комплексообразователя оказывает влияние на скорость массопереноса. Это происходит за счёт изменения потока миграции к поверхности электрода. Нейтральными считаются те соли, ионы которых не разряжаются на поверхности электрода. Для таких ионов скорость миграции равняется скорости диффузии. Поэтому они как бы неподвижны в электролите.
Помимо миграции на скорость доставки вещества к поверхности электрода сильное влияние оказывает конвекция. Любое конвективное движение жидкости приводит к уменьшению толщины диффузного слоя (не путать с диффузионным слоем в ДЭС) и возрастанию скорости процесса. На практике перемешивание электролита позволяет повысить предельную плотность тока в десятки раз.
Замедленность стадии диффузии наблюдается при электроосаждении из неперемешиваемых электролитов, которые содержат ионы таких металлов как свинец, олово, серебро, медь и ряда других, для которых характерны высокие токи обмена. Однако при интенсивном перемешивании раствора диффузионные ограничения могут быть сняты. Тогда проявляется замедленность стадии переноса электронов.
Поэтому замедленность стадии диффузии является не особенностью механизма реакции, а определяется характером проведения процесса.
1.6.2. Перенапряжение реакции переноса электрона
Теория, объясняющая зависимость тока от потенциала электрода или перенапряжения при замедленной стадии переноса электрона, называется теорией замедленного разряда. Здесь предполагается, что стадией, определяющей скорость реакции, является перенос электронов.
При равновесном потенциале (Е=Ер) скорость реакции восстановления (iK) равна скорости реакции окисления (ia). В этом случае происходит обмен ионами между раствором и электродом. Скорость этого обмена характеризуется током обмена i0. При сдвиге потенциала от равновесного значения (то есть при поляризации электрода) через электрод протекает внешний ток, равный i(0) = ia - ik. Анодные токи принято считать положительными, а катодные – отрицательными.
Соотношение между перенапряжением и плотностью тока называется уравнением Тафеля:
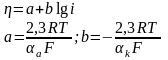 - константы.
- константы.
a и k – коэффициенты переноса анодной и катодной реакций. Коэффициент переноса характеризует степень влияния электрического поля электрода на энергию активации электрохимической стадии. Он определяет также симметрию катодного и анодного процессов. Для данной электрохимической реакции коэффициент переноса сравнительно мало изменяется при переходе от одного материала к другому. Чаще всего 0,5.
Катодная (1) и анодная (2) поляри-
зационные кривые в полулогариф-
мических координатах при замед-
ленной стадии переноса электронов
Рисунок (*)
Зависимость
перенапряжения от логарифма скорости
катодного или анодного процесса имеет
линейный характер (рис.(*)) только при
достаточно высоких перенапряжениях
(при
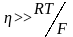 ).
При низких перенапряжениях надо
использовать полное уравнение для тока:
).
При низких перенапряжениях надо
использовать полное уравнение для тока:
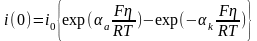 ,
где
,
где
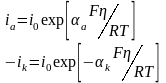
Выражение
i(0)
– это уравнение поляризационной кривой
при замедленной стадии переноса
электрона. Выражение для анодного тока
получают, когда
(при >0),
а для катодного, когда
 (при <0).
Выражения для a
и k
представляют
собой частные поляризационные кривые.
Они описывают истинную скорость анодного
или катодного процесса.
(при <0).
Выражения для a
и k
представляют
собой частные поляризационные кривые.
Они описывают истинную скорость анодного
или катодного процесса.
Соотношение между полной поляризационной кривой и частными кривыми представлено на рисунке (**).
|
Рис.** Поляризационные кривые при замед- ленной стадии переноса электронов: 1-для скорости анодного процесса (ia); 2-для скорости катодного про-са (iK); 3-суммарная кривая i(0) = ia - ik.
|
Аналогичный характер имеют и зависимости «плотность тока-потенциал».
Изменение концентрации компонента приводит к смещению поляризационной кривой.
Рис. ***Поляризационные кривые разря да-ионизации при различных концентрациях ионов металла в растворе: 1,2,3– катодные поляризационные кривые при концентрациях C1<C2<C3; 4–анодная поляризационная кривая
Из рисунка видно, что кривые нелинейны вблизи равновесного потенциала. Положение анодной кривой не зависит от концентрации ионов в растворе. Пересечение линейных участков катодных кривых с линейным участком анодной кривой происходит в точке со следующими координатами:
- по оси ординат соответствующих равновесному потенциалу,
- по оси абсцисс соответствующих логарифму тока обмена.
Лекция 4
2.4.4.3. Электроосаждение металлов из комплексных электролитов с предшествующими разряду химическими реакциями
При электроосаждении металлов из электролитов, содержащих комплексные ионы металла, возможен разряд различных сортов частиц. Это могут быть:
простые комплексные ионы металла;
комплексные ионы, преобладающие в растворе;
комплексные ионы, образующиеся в результате химической реакции диссоциации преобладающих комплексов.
В последнем случае стадии разряда предшествует стадия диссоциации преобладающего компонента. Такая, предшествующая разряду химическая реакция, может:
в первом случае, быть быстрой по сравнению со стадией разряда;
во втором случае, лимитировать скорость процесса в целом.
При электроосаждении металлов замедленность стадии, предшествующей химической реакции, наблюдается сравнительно редко. На её наличие обычно указывает низкое значение предельного тока (рис. – кривые 1 и 2).
Поляризационные кривые при за- медленной химической реакции диссоциации преобладающего комплекса:
3- высокая скорость диссоциации. |
Эти значения значительно меньше, чем предельный ток диффузии. Кроме того, для предельного тока химической реакции характерна независимость от перемешивания электролита.
Предшествующие химические реакции могут быть как гомогенными, то есть протекающими в объёме раствора, так и гетерогенными, протекающими на поверхности электрода. В последнем случае значение предельного тока зависит от способа подготовки или обработки электрода.
Перенапряжение, возникающее в результате замедленного протекания химической реакции, называется перенапряжением химической реакции. Оно связано с изменением концентрации реагирующего вещества у поверхности электрода. Перенапряжение диффузии и перенапряжение химической реакции возникают в результате изменения концентрации реагирующего вещества. Поэтому их называют концентрационным перенапряжением.
Поляризационные кривые при замедленности стадии химической реакции диссоциации аналогичны кривым, характерным для замедленной стадии диффузии.
На кривых, построенных в полулогарифмических координатах, в катодном процессе наблюдается предельный ток химической реакции.
При этом зависимость логарифма скорости анодного процесса от перенапряжения (или от потенциала электрода) линейна и описывается уравнением, аналогичным уравнению Тафеля.
2.4.4.4. Анодное растворение и пассивация металлов
Анодное растворение металлов является разновидностью электродных процессов и характеризуется свойственными этим процессам кинетическими зависимостями. Для протекания анодного растворения металла с заданной скоростью необходимо достаточно большое смещение его стационарного (равновесного) потенциала в положительную сторону, а также обеспечение интенсивного транспортирования реагирующих частиц в приэлектродной зоне.
Рассмотрим общие закономерности электролиза в средах с ионной проводимостью, которые применимы к анодному растворению. Пусть на электроде протекает только один электрохимический процесс, например, растворение металла. В этом случае процесс подчиняется законам Фарадея. Количественная сторона электродных процессов определяется первым и вторым законами Фарадея. Согласно первому закону, масса продуктов реакции q, образовавшихся на электродах, прямо пропорциональна силе тока I и времени его протекания t, то есть прямо пропорциональна количеству прошедшего электричества:
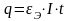 ,
(1)
,
(1)
где εЭ – ЭХ эквивалент, численно равный количеству вещества, выделяющегося при прохождении через систему одного кулона электричества.
Электрохимический эквивалент прямо пропорционален атомному весу металла и обратно пропорционален валентности, с которой он переходит в раствор. То есть, ЭХ эквивалент определяется природой металла и условиями процесса. Например, в зависимости от валентности хрома его ЭХ эквивалент колеблется от 0,973 (z = +2) до 0,323 г/(Ач) (при z = +6).
Электрохимический эквивалент сплава вычисляется по формуле:
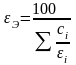 ,
(2)
,
(2)
где ci – процентное содержание i-го компонента в сплаве; I – электрохимический эквивалент i-го компонента.
Согласно второму закону, при прохождении через различные электролиты одного и того же количества электричества массы образовавшихся на электродах веществ (продуктов реакции) пропорциональны химическим эквивалентам этих веществ. То есть химический эквивалент εХ пропорционален электрохимическому эквиваленту:
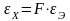 (3)
(3)
Подставив (1) в (3), получим:
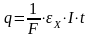 ,
(4)
,
(4)
где F = 96500 Кл – число Фарадея, равное количеству электричества, требующееся для растворения одного грамм-эквивалента металла.
На электроде при условии превышения определённого значения его потенциала могут происходить несколько электрохимических процессов одновременно. Например, кроме анодного растворения металла, его окисление, выделение кислорода и т.д. Поскольку для осуществления побочных реакций затрачивается определённая часть общего тока, говорят о величине выхода по току для основной реакции (растворения металла). Она определяется соотношением:
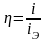 или
или
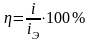 ,
,
где i – плотность тока, расходуемого на основную реакцию; iЭ – общая плотность тока, протекающего через электрод.
Необходимо отметить, что выход по току определяется аналогично и для катодных процессов. Для процессов осаждения металла побочной реакцией является реакция выделения водорода.
Выход по току не является постоянной величиной. Он зависит от материала электрода, состава электролита, величины поляризации электрода и других факторов. Например, выход по току для анодного растворения в хлоридном электролите колеблется от 10% при ЭХО молибдена до 99% - инструментальной стали. Наблюдаемые в некоторых случаях значения выхода по току, превышающие 100%, объясняются, как правило:
затруднениями определения истинной валентности растворяющегося металла (а, следовательно, и величиной его ЭХ эквивалента);
химическим растворением металла;
выпадением в раствор нерастворяющихся в полной мере компонентов сплава и уносом их с раствором.
Механизм анодного растворения металлов существенно влияет на скорость процесса, а также на качество обработанной поверхности. Рассмотрим закономерности различных случаев анодного растворения при изменении потенциала анодно поляризованного металла и других факторов процесса.

Рис.* Анодная поляризационная кривая
Рассмотрим следующие условия:
относительно малая величина анодной поляризации металла;
хорошая растворимость образующихся соединений;
отсутствие на поверхности электрода побочных процессов, приводящих к её пассивированию.
В этом случае происходит активное растворение (кривая АВ). К характерным особенностям активного растворения относится:
различная скорость растворения отдельных зёрен и кристаллографических плоскостей поверхности металла (так называемое структурное травление);
переход металла в раствор с низшей устойчивой валентностью (например, в случае железа равной +2);
обусловленность анодной поляризации замедленностью электрохимической реакции, то есть протекание процесса растворения в режиме электрохимической кинетики с тафелевской зависимостью скорости растворения от роста потенциала.
Логарифмическая зависимость поляризации электрода от плотности тока (уравнение Тафеля) имеет следствием весьма незначительное изменение потенциала при достаточно большом изменении скорости процесса. Поскольку в рассматриваемом случае анодного растворения на электроде происходит только одна реакция (переход катионов в раствор), выход по току для неё близок к единице.
Условия анодного растворения в активном режиме соблюдаются в большинстве случаев лишь для небольшого диапазона значений положительного потенциала растворяющегося металла. Уже при относительно малом перенапряжении параллельно начинаются побочные процессы, например, образование окислов. Это происходит по причине близости по величине значений равновесных потенциалов растворения и окисления многих металлов. С ещё бóльшей лёгкостью (при меньших потенциалах) на поверхности металла происходит образование адсорбционных слоёв, например, из кислорода. Эти слои экранируют поверхность частично или полностью и затрудняют анодное растворение металла.
Первоначальные пассивирующие слои лишь частично препятствуют перемещению через них катионов. Кроме того, они обладают невысокой стойкостью из-за воздействия активных анионов электролита и подкисления прианодного пространства из-за выделения кислорода. Полная пассивация металла наступает при более высоком потенциале пассивации п (или Фладе-потенциале). Величина этого потенциала зависит от ионного состава электролита, его рН, температуры, скорости прокачивания или перемешивания электролита (если процесс определяют концентрационные изменения в приэлектродном пространстве). Присутствие в растворе активирующих ионов, подкисление его, нагрев затрудняют образование экранирующих слоёв.
Величину плотности тока, соответствующей потенциалу п, называют плотностью тока пассивации iп. Присутствие в растворе активных ионов увеличивает плотность тока пассивации в несколько раз. Нагревание электролита существенно повышает значения iп и п. Небольшое превышение потенциалом величины п (точка В на рисунке*) при отсутствии или малой концентрации активных ионов в электролите вызывает резкое понижение анодной плотности тока (точка С) до значений порядка 10-3-10-7 А/см2. Это связано с образованием на поверхности электрода стойких пассивирующих слоёв, как правило, окисной фазовой природы. Значительная часть протекающего через электрод тока затрачивается на увеличение пассивирующей плёнки. Дальнейшее повышение потенциала практически не влияет на величину тока, соответствующую пассивной области поляризационной кривой.
Свойства окисных слоёв, образующихся на поверхности металла в режиме пассивного растворения, определяются природой металла и условиями протекания процесса. Толщина плёнки зависит от состава металла, возрастая с увеличением положительного потенциала и повышением рН раствора. Толщина плёнки может колебаться от 20 Å на железе и до 5000 Å на титане.
В зависимости от величины анодного потенциала состав пассивирующей плёнки существенно меняется. С его ростом повышается валентность металла в окисных соединениях. То есть металл как бы доокисляется. Части плёнки, соприкасающиеся с электролитом, более насыщены кислородом, чем внутренние слои.
Внешняя и внутренняя области пассивирующей окисной плёнки имеют различный тип проводимости. В области, пограничной с раствором, создаётся дырочная проводимость (р-типа), а около металла – электронная проводимость (п-типа). Средний слой с приблизительно постоянным составом обладает проводимостью i-типа. (Проводимость i-типа, это собственная проводимость, при которой проводниками являются ионы. Однако, их количество незначительно). Таким образом, окисная плёнка эквивалентна полупроводниковому диоду с односторонней проводимостью. Ток, протекающий через плёнку, представляет собой сумму электронного и ионного токов. Соотношение этих токов влияет на величину выхода по току. Если электронная проводимость пассивной плёнки значительно больше ионной, то это способствует возникновению в ней диффузионного потенциала, аналогичного потенциалу в растворах.
Как правило, растворение металлов в пассивной области (участок CD на анодной поляризационной кривой) происходит при образовании катионов высшей валентности (например, Fe+3, Cr+6). Гидродинамические условия не влияют на кинетику растворения металла в рассматриваемой пассивной области. Это связано с тем, что при столь малой интенсивности растворения мала вероятность развития диффузионных ограничений процесса. Пассивационные явления на поверхности анодно растворяющегося металла чрезвычайно важны для процессов ЭХО. Они существенно влияют на скорость процесса и качество обработанной поверхности.
Рассмотрим плёночную и адсорбционную теорию пассивности.
Плёночная теория пассивности объясняет состояние повышенной электрохимической устойчивости металлов образованием на их поверхности очень тонкой защитной плёнки из нерастворимых продуктов взаимодействия металла со средой. Плёнка состоит обычно из одной фазы, может быть солевой, гидроокисной или (наиболее часто) окисной природы. Поведение металла в пассивном состоянии определяется не свойствами самого металла, а физико-химическими свойствами плёнки. При анодной поляризации металла в кислородосодержащем электролите плёнка находится в динамическом равновесии с раствором. То есть растворение внешней части плёнки под химическим воздействием электролита компенсируется одновременным процессом анодного возобновления плёнки.
Плёночная теория не лишена ряда недостатков. Она не в состоянии объяснить ряд экспериментальных данных. Например:
окисные слои, осаждающиеся на поверхности металла, имеют рыхлую структуру и не могут оказывать изолирующего и пассивирующего действия;
в некоторых случаях пассивность наступает внезапно; этого не должно быть, если бы пассивация наступала в результате роста окисного изолирующего слоя на поверхности металла;
отдельные металлы при пассивации вообще не покрываются плёнками, так как продукты их окисления хорошо растворимы;
трудно с позиций плёночной теории объяснить большую скорость прохождения ионов через пассивирующий слой, так как обычно такая диффузия очень медленна.
Адсорбционная теория главную роль в возникновении пассивного состояния металла отводит образованию на его поверхности более тонких защитных адсорбционных слоёв молекулярного, атомарного и отрицательно ионизированного кислорода, а также гидроксильных ионов. Причём адсорбированные частицы образуют монослой или долю его. Процесс образования адсорбционного пассивирующего слоя может проходить одновременно с анодным растворением металла.
Существует два варианта объяснения адсорбционного механизма пассивности – химический и электрохимический. Согласно химическому варианту адсорбированный кислород насыщает активные валентности поверхностных атомов металла, уменьшая их химическую активность. Электрохимический вариант объясняет возникновение пассивности электрохимическим торможением анодного процесса растворения. Образовавшиеся адсорбционные слои, изменяя строение двойного слоя и смещая потенциал металла к положительным значениям, повышают работу выхода катиона в раствор. В результате этого растворение металла затормаживается.
Теории пассивности дополняют друг друга. Лишь их сочетание наиболее полно объяснить все наблюдаемые проявления пассивности.
Допустим, что в электролите содержится значительное количество активирующих ионов, например ионов галогенов. Увеличение потенциала электрода до значения, равного потенциалу активации φакт, приводит к резкому возрастанию плотности тока и установлению состояния анодной активации (области YK и Y’K’ на рисунке*). Относительная активность анионов различной природы, их способность активировать металл, в частности железо, характеризуется следующим радом:
Cl¯ > Br¯ > I¯ > F¯ > ClO4¯ > SO4¯
Потенциал активации смещается к более отрицательным значениям при:
увеличении концентрации в растворе активирующих анионов;
увеличении температуры электролита;
подкислении электролита.
Эти факторы затрудняют пассивацию металла и облегчают растворение.
Анодная активация металла позволяет получить довольно высокие скорости растворения металлов при сравнительно низкой величине анодной поляризации. Предельная плотность тока активированного растворения (точки К и К´ на рисунке*) при естественном движении электролита составляет 1-2 А/см2. При интенсивном движении она увеличивается в десятки раз.
При достаточно высокой положительной поляризации запассивированного металла в электролите, не содержащем активирующих ионов, наблюдается перепассивация или переход в транспассивное состояние. При этом скорость растворения металла заметно увеличивается. Этому состоянию соответствует кривая DE на рисунке*. Нерастворимые ранее окислы при достижении потенциалов перепассивации могут переходить в окислы более высокой валентности. Эти окислы в данных условиях обладают растворимостью, а значит, низкими защитными свойствами.
Во многих случаях растворение в транспассивном состоянии возможно лишь при потенциалах более положительных, чем потенциал выделения кислорода (точка G на рисунке *). В результате на электроде происходят два параллельных процесса.