

Тема: «адсорбция»
Явление поглощения одним веществом других веществ называется сорбцией. Сорбция в толще поглотителя называется абсорбцией. Повышение концентрации газообразного или растворенного вещества на поверхности раздела фаз, например, на поверхности раздела твердое тело – газ, твердое тело – раствор, жидкость – газ (воздух) называется адсорбцией. Газ или растворенное вещество, концентрирующееся на поверхности, называется адсорбтивом, а вещество, на поверхности которого происходит адсорбция – адсорбентом.
Различают физическую адсорбцию, когда частицы адсорбированного вещества не образуют химических связей с адсорбентом, и хемосорбцию, когда адсорбционные силы имеют химическую природу.
Адсорбция определяется природой поглощаемого вещества и поглотителя, ее величина зависит от температуры, давления поглощаемого газа, концентрации раствора, из которого осуществляется адсорбция.
Количество адсорбированного вещества пропорционально площади поверхности тела. Поэтому вещества в мелкораздробленном состоянии, имея большую поверхность, обладают значительной адсорбционной способностью. К эффективным адсорбентам можно отнести уголь, силикагель, глину, каолин, а также целлюлозу, фильтровальную бумагу, хлопчатобумажную ткань, натуральный шелк, шерсть и другие материалы.
Уравнения, описывающие Адсорбцию
Фундаментальное уравнение Гиббса
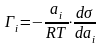 ,
,
где Г – избыточное количество адсорбата в поверхностном слое по сравнению с объемной фазой (моль/м2), ai – активность i-го компонента в состоянии равновесия, Т – температура (К), R – универсальная газовая постоянная, dσ – бесконечно малое изменение поверхностного натяжения при бесконечно малом изменении равновесной активности (dai).
Для разбавленных растворов активность равна концентрации, и уравнение принимает вид:
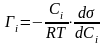 .
.
Данное уравнение является универсальным, так как справедливо для всех типов адсорбционных процессов. Однако область практического применения ограничивается системами, для которых доступно экспериментальное измерение поверхностного натяжение (жидкость – жидкость и жидкость - газ)
В упрощенном виде, когда речь идет о конечном интервале концентраций, данное уравнение можно записать следующим образом:
 .
.
Вещества, для которых Г > 0, снижают поверхностное натяжение и называются поверхностно-активными веществами (ПАВ). Вещества, для которых Г < 0, повышают поверхностное натяжение и называются поверхностно-инактивными веществами (ПИВ). Вещества, не влияющие на поверхностное натяжение, называю поверхностно-неактивными (ПНАВ).

Как правило, ПАВ являются дифильными, то есть их молекулы обладают полярной (гидрофильной) и неполярной (гидрофобной) частями. В случае органических молекул неполярные части обычно имеют углеводородную структуру, а полярные - электрофильные атомы ( О, N, S, P и т.д.)

Зависимость поверхностного натяжения от концентрации ПАВ описывается эмпирическим уравнение Шишковского: σ=σ0-В∙ln(1+А∙С), где В и А – эмпирические константы, С – концентрация ПАВ, σ и σ0 – поверхностное натяжение раствора и растворителя. Данное правило применимо для ПАВ с не очень длинным углеводородным радикалом и слабо диссоциированными гидрофильными группами.
Способность веществ влиять на величину поверхностного натяжения характеризуется поверхностной активностью (g) (при p, t = const):
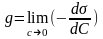
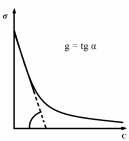
По способности к диссоциации в водных средах ПАВ классифицируют:

Все дифильные поверхностно-активные вещества в зависимости от их способности к самоорганизации в растворах (способности к образованию надмолекулярных структур) делят на две большие группы: истинно растворимые и коллоидные (мицеллообразующие).
К ПАВ первой группы относятся вещества дифильной структуры, но содержащие короткий углеводородный радикал, хорошо растворимые в воде и образующие молекулярно-дисперсные (истинные) растворы вплоть до концентраций, соответствующих их насыщению и разделению системы на две макрофазы. Это низкомолекулярные члены гомологических рядов органических кислот, спиртов, аминов, эфиров и др. дифильных веществ. ПАВ этой группы находят применение в качестве смачивателей, пенообразователей и гидрофобизаторов при флотации, диспергаторов (веществ, облегчающих образование новых поверхностей) и т.д.
Значительно больший интерес в прикладном и научном отношении имеют вещества второй группы, главной особенностью которых является способность образовывать термодинамически устойчивые (лиофильные) дисперсные системы – ассоциативные, или мицеллярные, коллоиды при концентрации выше некоторого критического значения, называемого критической концентрацией мицеллообразования (ККМ). При ассоциации лиофильные части молекул ПАВ (имеющие большое сродство к растворителю) располагаются на периферии мицеллы, внутри ее находятся лиофобные части молекул. Так, в водных растворах неполярные углеводородные радикалы молекул ПАВ образуют ядро мицеллы, а полярные группы обращены к воде, такие мицеллы называются прямыми (а). В неполярных средах образуются обратные мицеллы, т.е. внутри мицеллы располагаются полярные группы (б).
Схематичное изображение мицеллы ПАВ в воде(а) и в неполярной среде (б)

Способностью к мицеллообразованию, в подавляющем большинстве случаев, обладают ПАВ, содержащие от 8 до 18 атомов углерода в углеводородной цепи (R).
Образование мицелл уменьшает энергию Гиббса G благодаря слипанию углеводородных радикалов и формированию частиц, которые можно трактовать как неполярные «капли» масла, покрытые оболочкой из гидрофильных групп. Чем длиннее углеводородная цепь, тем больше выигрыш энергии за счет выхода цепей из полярной среды и когезии их в мицелле и тем меньше концентрация ПАВ, необходимая для образования мицелл. Эта закономерность проявляет себя в резком уменьшении величины ККМ в гомологических рядах ПАВ по мере увеличения молекулярной массы. В гомологических рядах ПАВ величина ККМ изменяется обратно пропорционально поверхностной активности (g), так что отношение ККМ соседних гомологов соответствует коэффициенту правила Дюкло–Траубе:
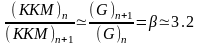
При равном количестве углеродных атомов величина ККМ возрастает:
1) при циклизации цепи (алкилсульфонаты → алкилбензолсульфонаты);
2) при введении в углеводородный радикал полярных групп, гетероатомов (стеарат → олеат → рицинолеат, миристат → дециловый эфир β-оксипропионата калия).
Таким образом, ароматические циклы, кратные связи, полярные группы, гетероатомы приводят к уменьшению гидрофобности углеводородного радикала.
Уравнение Ленгмюра
Уравнение Ленгмюра справедливо для широкого интервала концентраций и для различных границ раздела фаз, как подвижных, так и неподвижных (твердое – жидкость, твердое – газ, жидкость – жидкость, жидкость-газ). Однако, данное уравнение выведено исходя из следующих положений:
Адсорбция локализована на отдельных энергетически активны центрах, каждый из которых может взаимодействовать только с одной молекулой адсорбтива (адсорбата). В результате может образоваться только мономолекулярный адсорбционный слой;
Адсорбционные силы действуют на малых расстояниях,
Адсорбированные молекулы не взаимодействуют друг с другом;
Поверхность адсорбента ограничена и по мере увеличения концентрации адсорбтива (адсорбата) происходит насыщение поверхности;
Адсорбированные молекулы могут покидать поверхность, а их место могут занимать другие молекулы
Зависимость адсорбции от давления газа (его концентрации) или содержания адсорбируемого вещества в растворе при данной температуре может выражаться уравнением адсорбции Лэнгмюра (изотермой Лэнгмюра)
Для
адсорбции из растворов

Для
адсорбции газов
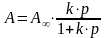
где А и А - количества адсорбированного вещества на единице поверхности адсорбента, выражаются в моль/м2, А - адсорбция в состоянии адсорбционного равновесия, А - максимально возможная адсорбция; С - концентрация (моль/л) адсорбируемого вещества в растворе в состоянии адсорбционного равновесия (равновесная концентрация); р - давление газа в состоянии адсорбционного равновесия (Па), К – константа Лэнгмюра. В общем случае А больше гиббсовского поверхностного избытка (Г), однако для ПАВ можно пренебречь концентрацией в объеме фазы по сравнению с очень высокой концентрацией в поверхностном слое и принять А≈Г.
Вид изотермы адсорбции Лэнгмюра

Изотерму адсорбции можно разделить на три области :
Область малых концентраций (давлений)
k<<1,
 -
уравнение
Ленгмюра сводится к уравнению изотермы
Генри
-
уравнение
Ленгмюра сводится к уравнению изотермы
Генри
Область средних концентраций (давлений)
Область высоких концентраций (давлений)
k
>>1,

Уравнение Фрейндлиха
Для газов, растворов неэлектролитов и слабых электролитов кривая изотермы адсорбции на твердом адсорбенте имеет вид параболы.
В некотором интервале концентраций, для не слишком разбавленных, но и не очень концентрированных растворов, адсорбция может быть описана эмпирическим уравнением Фрейндлиха:
для
адсорбции из раствора
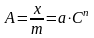 ,
,
для
адсорбции газа

где А - величина адсорбции (моль/г), х - количество растворенного вещества, адсорбированного массой m поглотителя и находящегося в равновесии с раствором концентрации С, р – давление газа в состоянии адсорбционного равновесия, а и n - эмпирические константы, характерные для данного процесса адсорбции в определенных пределах, n характеризует кривизну изотермы адсорбции, обычно имеет значения от 0,1 до 0,6.
Вид изотермы адсорбции Фрейндлиха

Строго говоря, количество адсорбированного вещества следует относить не к единице массы, а к единице площади поверхности, но для мелкораздробленных веществ и однородных суспензий (например, взмученный порошок активированного угля) поверхность растет пропорционально обшей массе.
Для нахождения значений эмпирических констант а и n по экспериментальным данным нужно прологарифмировать уравнение, тогда оно примет вид:
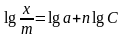 .
.
Задачи для самостоятельного решения:
На уравнение Гиббса
При уменьшении концентрации раствора новокаина с 0,2 моль/л до 0,15 моль/л поверхностное натяжение возросло с 69∙10-3 Н/м до 71∙10-3 Н/м. Рассчитайте величину адсорбции в данном интервале концентраций (Т=298К).
Рассчитайте величину адсорбции масляной кислот при 50⁰С в данном интервале концентраций если известно, что при увеличении концентрации масляной кислоты от 0.125 моль/л до 0,25 моль/л поверхностное натяжение понизилось с 55,1∙10-3 Н/м до 47,9∙10-3 Н/м
На уравнении Фрейндлиха
Вычислить величину адсорбции аргона на угле при -78,3ºС в м3/кг (приведенных к нормальным условиям), если давление аргона равно 1,01∙105 Па, a=4,83∙10-5 м3/кг, n=1,66.
Определите адсорбцию уксусной кислоты на животном угле при 25С, если равновесная молярная концентрация кислоты равна: а) 0,062 моль/л, б) 0,882 моль/л. Константы a=0,155 моль/кг, а n=2,38.
Для адсорбции уксусной кислоты на адсорбенте константы уравнения Фрейндлиха равны: a=0,50 моль/г, n=0,45. Рассчитайте величину адсорбции при равновесной концентрации 0,22 моль/л.
Раствор уксусной кислоты объемом 30 мл с концентрацией 0,05 моль/л взболтали с 1 г адсорбента. После достижения равновесия пробу оттитровали раствором гидроксида натрия (С=0,05 моль/л). На титрование затрачено 12 мл титранта. Вычислите величину адсорбции уксусной кислоты.
Раствор пропионовой кислоты объемом 50 мл с концентрацией 0,2 моль/л взбалтывался с адсорбентом массой 2 г. После достижения адсорбционного равновесия на титрование фильтрата объемом 10 мл был затрачен титрант объемом 15 мл С(KOH)=0,05 моль/л. Определите величину адсорбции уксусной кислоты.
На уравнение Ленгмюра
Рассчитайте адсорбцию азота (в м3/кг) на слюде при 90 К по уравнению Лэнгмюра, если давление азота равно 1580 , Г∞= 0,04 м3/кг, и k =1,18∙10-3
Рассчитайте адсорбцию азота (в м3/кг) на слюде при 90 К по уравнению Лэнгмюра, если давление азота равно 477 Па, Г∞= 0,04 м3/кг, k =1,18∙10-3.
Экспериментально установлено, что максимальная величина адсорбции ПАВ (Mr=60) некоторым адсорбентом составляет 5,0∙10-3 моль/г; k=16,6 моль/л. Сколько граммов вещества адсорбировалось из раствора с равновесной концентрацией 0,1 моль/л двумя граммами данного сорбента?
ТЕМА: «ДИСПЕРСНЫЕ СИСТЕМЫ. КЛАССИФИКАЦИЯ, МЕТОДЫ ПОЛУЧЕНИЯ И ОПТИЧЕСКИЕ СВОЙСТВА»
Дисперсной называется двух- или многофазная, т.е. гетерогенная система, в которой, по крайней мере, одна из фаз представлена очень маленькими частицами, размеры которых тем не менее заметно превосходят молекулярные. Частицы раздробленного вещества при этом называются дисперсной фазой, а гомогенная фаза, в которой они распределены (растворитель), представляет собой дисперсионную среду. Примерами дисперсных систем могут служить дымы, туманы, взвеси различных веществ, например глины в воде и т.д.
Дисперсные системы классифицируют по агрегатному состоянию дисперсной фазы и дисперсионной среды (Таблица). Системы, представляющие собой мелкие капельки жидкости в жидкой дисперсионной среде, называются эмульсиями. Примеры эмульсий молоко, латексы, различные косметические препараты и т.д. Высокодисперсные системы с твердой дисперсной фазой и жидкой дисперсионной средой называют золями при невысокой концентрации дисперсной фазы (когда частицы обособлены) и гелями, когда частицы агрегируют и образуют связно дисперсную систему. В случае грубых дисперсий твердого вещества в жидкой дисперсионной фазе различают малоконцентрированные суспензии и высококонцентрированные пасты.
Дисперсные системы различаются по величине частиц раздробленного вещества, или, как говорят, по степени дисперсности. Различают грубодисперсные и коллоидно-дисперсные системы. В суспензиях и эмульсиях радиус частиц больше 0,1 мк. Такие дисперсные системы, как, например, взвесь глины в воде, не обладают осмотическим давлением, не фильтруются через бумажные фильтры и являются неустойчивыми, т.е. не остаются долго во взвешенном состоянии в жидкой среде, а выпадают под действием силы тяжести на дно сосуда. Это кинетически неустойчивые системы. С повышением степени дисперсности, т.е. уменьшением частиц дисперсной фазы, системы приобретают новые качества. Так, дисперсные системы, состоящие из частиц с радиусом от 0,1 мк до 1 ммк, качественно отличны от суспензий. Частицы таких систем находятся в непрерывном хаотическом движении, вследствие чего обладают и осмотическим давлением, и способностью к диффузии. Благодаря непрерывному движению частиц они остаются кинетически устойчивыми. Такие системы в проходящем свете прозрачны, в то время как суспензии мутны. Подобного рода дисперсные системы часто называют коллоидными растворами. Частицы коллоидных растворов (золей) значительно больше молекул, так как они представляют собой агрегаты из достаточно большого числа молекул или атомов.
В суспензиях частицы можно наблюдать при помощи микроскопа и тем самым обнаружить поверхность раздела фаз. Коллоидные частицы из-за незначительных размеров нельзя обнаружить даже под микроскопом.
В истинных растворах размер частиц доведен до размеров молекул или ионов. В них нет поверхности раздела между составляющими компонентами, т.е. растворителем и растворенным веществом. Истинные растворы агрегативно устойчивы.
Таблица
Классификация дисперсных систем по агрегатному состоянию


Коллоидные системы по термодинамической устойчивости делятся на лиофобные и лиофильные. Термодинамически устойчивые системы, образующиеся при самопроизвольном диспергировании одной из фаз, называются лиофильными. Самопроизвольному диспергированию способствует усиление взаимодействия дисперсной фазы с дисперсионной средой. Примерами лиофильных коллоидных систем могут служить растворы мыл и других поверхностно-активных веществ. К ним также относят растворы белков и других высокомолекулярных соединений.
Основным свойством лиофобных систем является их термодинамическая неустойчивость, связанная с большим запасом свободной поверхностной энергии на развитой межфазной поверхности раздела. Самопроизвольно из макроскопических фаз они не образуются. Получение их требует затраты внешней энергии – механической (растирание), химической (проведение химических реакций), электрической (распыление под действием электрического тока). Однако такие коллоидные системы могут быть кинетически устойчивыми, т.е. сохраняться без изменений в течение более или менее длительного времени.
Существуют две группы методов получения лиофобных коллоидных систем. Методы, основанные на раздроблении крупных частиц на более мелкие, получили название методов диспергирования. Методы, связанные с агрегацией молекул или ионов в более крупные частицы, называются конденсационными. Важное условие для получения устойчивой коллоидной системы – присутствие стабилизаторов, т.е. веществ, которые, адсорбируясь на поверхности коллоидных частиц, создавали бы достаточно интенсивное взаимодействие между поверхностью и окружающей средой (растворителем).
К методам конденсации относятся следующие способы получения коллоидных систем.
Конденсация молекул испаряющегося вещества, соединяющихся в мелкие частицы.
Замена растворителя, т.е. такое изменение среды, при котором вещество из растворимого становится нерастворимым.
Химические реакции в растворе, сопровождающиеся образованием трудно растворимых веществ.
Однако во всех этих случаях коллоидные системы получаются тогда, когда растворимость дисперсной фазы ничтожна мала. При несоблюдении этого условия возможно образование молекулярных растворов. Кроме того, необходимо, чтобы между частицами и средой существовало взаимодействие, препятствующее связыванию частиц друг и другом.

В целом рассматриваемая коллоидная частица (гранула) несет некоторый положительный заряд. При сближении двух таких частиц, несущих на поверхности одноименный заряд, будет происходить отталкивание, препятствующее их слипанию (электростатический барьер). Сближению коллоидных частиц препятствует также и слой молекул растворителя, в частности воды, входящих в сольватную (гидратную) оболочку ионов на поверхности частицы. Кроме того, на поверхности коллоидных частиц могут адсорбироваться молекулы специально добавляемых веществ-стабилизаторов (адсорбционно-сольватный барьер).
Наличие электростатического и адсорбционно-сольватного барьера, препятствующих агрегации (соединению) частиц, обеспечивают агрегативную устойчивость лиофобным коллоидным системам, т.е. такие системы не изменяются заметно в течение длительного времени (иногда десятилетиями), несмотря на термодинамическую неустойчивость.
Так как лиофобные дисперсные системы являются термодинамически неравновесными, в них могут идти процессы укрупнения частиц и соответственно уменьшения межфазной поверхности. Наиболее характерный и общий для дисперсных систем путь перехода к равновесному состоянию – коагуляция, т.е. слипание частиц дисперсной фазы. Часто коагуляция сопровождается появлением мути, изменением окраски коллоидных растворов, образованием осадка (явная коагуляция). Когда происходит укрупнение частиц без видимых внешних изменений, говорят о скрытой коагуляции.
Коагуляция может происходить при действии на систему различных факторов: механическое воздействие (перемешивание или встряхивание), резкое охлаждение или нагревание, пропускание электрического тока. Иногда коагуляция может произойти в результате «старения» или химических изменений, происходящих в золе.
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА КОЛЛОИДНЫХ СИСТЕМ
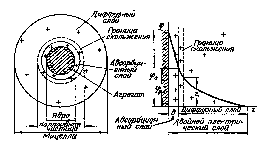
На границе раздела твердой фазы и раствора, как правило, возникает двойной электрический слой (ДЭС). Его происхождение может быть двояким. Во-первых, возможна ионизация молекул, составляющих поверхностный слой твердой фазы, например, ионизация молекул H2SiO3, образующихся на поверхности SiO2 в воде. Во-вторых, на поверхности твердой фазы может происходить адсорбция одного из ионов, присутствующего в растворе электролита. При этом на поверхности преимущественно адсорбируются ионы, входящие в состав твердой фазы или близкие к ним по природе. Так на поверхности частиц золя AgCl будут адсорбироваться ионы Ag+ или Cl– в зависимости от того, какие из них имеются в растворе в избытке. Ионы, определяющие заряд коллоидной частицы (гранулы) называются потенциалобразующими. К заряженной поверхности частиц будут притягиваться ионы противоположного знака, т.е. противоионы, образуя двойной электрический слой.
Двойной электрический слой на поверхности коллоидных частиц включает так называемый адсорбционный слой и диффузный слой. Адсорбционный слой образован частью противоионов, которые прочно связаны с ядром мицеллы электростатическими силами (притягивание разноименных зарядов) и адсорбционными силами. Остальные противоионы, благодаря тепловому движению и взаимному отталкиванию, уходят на некоторое расстояние от межфазной границы, образуя диффузный слой ионов, который удерживается у поверхности только электростатическими силами (см. рис.).
Каждая точка электрического поля двойного слоя, образованного потенциалопределяющими ионами и противоионами, характеризуется определенным значением электрического потенциала. Причем в адсорбционном слое, т.е. на малых расстояниях от поверхности, падение потенциала происходит круто, а далее в диффузном слое более полого.
Потенциал двойного слоя, отвечающий границе скольжения при движении дисперсной фазы и дисперсионной среды относительно друг друга, называется электрокинетическим или -потенциалом (дзета-потенциалом). Место границы скольжения определяется действием адсорбционных и электрических сил, а также свойствами раствора, окружающего частицы, в частности, вязкостью прилегающих слоев жидкости. Граница скольжения может совпадать с границей между адсорбционными и диффузным слоями или находиться несколько дальше от поверхности, где-то в диффузном слое.
Строение
мицеллы и двойного электрического
слоя.



Электрофорез находит широкое применение в медико-биологических исследованиях. В клинической практике электрофоретические методы применяются для диагностики многих заболеваний, для разделения аминокислот, нуклеиновых кислот, антибиотиков, ферментов, антител, для определения чистоты белковых препаратов и т.д.




Дзета-потенциал является важной характеристикой коллоидных систем. Во многих случаях отмечается закономерность: чем больше величина -потенциала, тем выше устойчивость золя. При значениях -потенциала ниже 0,03 В (критический потенциал) наступает коагуляция золя.

При добавлении раствора электролита к золю противоионы нейтрализуют заряд на поверхности коллоидной частицы (происходит сжатие ДЭС), что позволяет частицам золя легче приближаться друг к другу, и это воздействие тем сильнее, чем больший заряд несет противоион.
Коагулирующий ион несет заряд, противоположный заряду коллоидной частицы, при этом порог коагуляции тем меньше, чем больше заряд (валентность) коагулирующего иона – правило Шульце-Гарди. Порогом коагуляции называется минимальная концентрация электролита, вызывающая коагуляцию.
Порог
коагуляции выражается в миллимолях
электролита на литр коллоидного раствора
(ммоль/л). Порог коагуляции вычисляют
по формуле: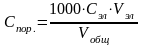 ,
,
где Сэл – концентрация раствора добавленного электролита, моль/л, Vэл – наименьшее число миллилитров раствора электролита, достаточное для коагуляции золя, Vобщ – суммарный объем золя после добавления электролита, мл.
При добавлении некоторых веществ нередко наблюдается повышение устойчивости лиофобных золей к коагулирующему действию электролитов. Такое стабилизирующее действие на дисперсные системы называется коллоидной защитой. Защитными свойствами обладают белковые вещества (желатин, альбумины, казеин), полисахариды (крахмал, декстрин), некоторые поверхностно-активные вещества. Если, например, к золю гидроксида железа (III) добавить некоторое количество желатина, то для коагуляции такого золя требуется значительно больше электролита, чем для коагуляции незащищенного золя. Коллоидную защиту объясняют адсорбцией стабилиаторов на поверхности частиц дисперсной фазы и образованием слоя.
Коллоидная защита широко используется при получении устойчивых лиофобных золей, применяемых в качестве лекарственных препаратов. Например, колларгол и протаргол содержат 8 и 70% высокодисперсного металлического серебра, стабилизированного гидролизатами белков.Коллоидная защита играет существенную роль в физиологических процессах. Содержание карбоната и фосфата кальция в крови значительно превышает их растворимость в воде. Отложению этих солей препятствуют защитные вещества крови, которые не позволяют коллоидным частицам нерастворимых солей объединяться в крупные агрегаты и осаждаться. Образование желчных и мочевых камней в организме связано с уменьшением при патологических состояниях защитного действия определенных веществ по отношению к билирубину, холестерину и уратам.
Задачи для самостоятельного решения:
Напишите формулу мицеллы для золя иодида серебра, полученного при добавлении к раствору KI объемом 30 мл с концентрацией 0,006 моль/л раствор AgNO3 объемом 40 мл с концентрацией 0,004 моль/л. Назовите части мицеллы.
Напишите формулу мицеллы для золя, полученного смешением 20 мл 0,01 моль/л раствора сульфата натрия и 50 мл 0,004 моль/л раствора хлорида бария.
Напишите формулу мицеллы для золя, полученного смешением 30 мл 0,01 моль/л раствора сульфата натрия и 20 мл 0,04 моль/л раствора хлорида бария.
Коагуляция золя сульфида золота объемом 650 мл наступила при добавлении раствора сульфата хрома (III) объемом 1,18 мл с концентрацией 0,025 моль/л. Вычислите порог коагуляции золя сульфат-ионами.
Золь бромистого серебра получен путем смешивания равных объемов 0,008 н раствора бромистого калия и 0,0096 н раствора азотнокислого серебра. Определить знак заряда частиц золя и написать формулу его мицеллы.
Коагуляция гидроксида алюминия объемом 2 л наступила при добавлении раствора гексацианоферрата (II) калия объемом 10,6 мл с концентрацией 0,01 моль/л. Вычислите порог коагуляции золя гексацианоферрат (II)-ионами.