

I Термохимия.
1) Термодинамические системы (открытая, закрытая, изолированная). Параметры и функции состояния системы. Энтальпия, энтропия, энергия Гиббса. |
Термодинамическая система – тело или совокупность тел, мысленно выделенная из окружающей среды. Открытая – обменивается с окружающей средой веществом и энергией. Закрытая – только энергией, изолированная – ни веществом, ни энергией. Параметры состояния системы – T,P,V, концентрация и т.д. Функции системы: внутренняя энергия U, энтальпия H, энтропия S, энергия Гиббса G.(функции, т.к. зависят от параметров системы).Тепловой эффект в ходе реакции – изменение энтальпии или энтальпия процесса. dH=dU+PdV. S – мера неупорядоченности системы(количественная мера беспорядка). Возможность протекания реакции характеризуется изменением энтропии. S=klnW, где k – пост. Больцмана, W-число микросостояний в макросистеме. Энергия Гиббса – изобарно-изотермический потенциал. dG=dH-TdS. dG характеризует возможность самопроизвольного протекания процесса.dG<0 – протекает, >0, самопроизвольно не протекает.
2) Тепловые эффекты химических реакций. Эндо- и экзотермические процессы. Закон Гесса и его следствия. Стандартная энтальпия образования вещества. |
3) Закон Гесса. Следствия закона Гесса: связь энтальпии химической реакции со стандартными энтальпиями образования, сгорания или разрыва связей для исходных веществ и продуктов реакции. Стандартные энтальпии образования и сгорания химических соединений. |
4) Закон Гесса для теплового эффекта физико-химического процесса. Следствия закона Гесса: связь энтальпии химической реакции со стандартными энтальпиями образования, сгорания или разрыва связей для исходных веществ и продуктов реакции.|
5) Стандартные энтальпии термодинамических процессов: энтальпия образования, сгорании. Энтальпии растворения, нейтрализации. Энергия связи. |
Тепловой эффект химической реакции или изменение энтальпии системы вследствие протекания химической реакции — отнесенное к изменению химической переменной количество теплоты, полученное системой, в которой прошла химическая реакция и продукты реакции приняли температуру реагентов. Стандартная энтальпия образования dH0обр – тепловой эффект реакции (ТЭР) образования одного моля в-ва из простых в-в, его составляющих, находящихся в устойчивых стандартных состояниях. dH0обрf простого в-ва = 0(относится к агрегатному состоянию, устойчивому при T=298K). Стандартная энтальпия сгорания dH0сгор – ТЭР сгорания одного моля в-ва до высших оксидов. Стандартная энтальпия разрыва связи(называется также Есв ) dH0св – энергия, поглощаемая при разрыве 1 моля связей двух атомов находящихся в газообразном состоянии при 298К. Энтальпия растворения складывается из теплоты разрушения кристаллической решетки (dH0реш >0) и теплоты гидратации(сольватации для неводных растворов) (dH0гидр <0). Энтальпия растворения может иметь как +, так и -. Стандартная энтальпия нейтрализации dH0нейтр – энтальпия реакции взаимодействия сильных кислот и оснований с образованием 1 моля воды. Стандартная энтальпия реакции dH0 – тепловой эффект реакции определённого числа молей реагентов задаваемого уравнением реакции при стандартных условиях. Экзотермическая реакция – реакция, сопровождающаяся выделением теплоты (dH <0). Эндо – поглощением (dH0 >0). Закон Гесса – Тепловой эффект(энтальпия) реакции процесса зависит только от начального и конечного состояния системы и не зависит от пути перехода её из 1 состояния в другое. Следствия: 1)энтальпия реакции равна разности сумм энтальпий образования конечных и начальных участников реакции с учётом их стехиометрических коэффициентов (домножение суммы на коэффициент). dH0реак = ΣdH0обр кон -ΣdH0обр нач. 2)Энтальпия реакции равна разности сумм энтальпий сгорания начальных и конечных реагентов с учётом их стехиометрических коэффициентов. dH0реак = ΣdH0сгор кон -ΣdH0сгор нач.3) Энтальпия реакции равна разности сумм энергий связей Eсв исходных и конечных веществ с учётом их стехиометрических коэффициентов. dHреак = ΣЕисх –ΣЕпрод.4) Энтальпия образования в-ва равна энтальпии разложения его до простых в-в с обратным знаком. 5) Энтальпия гидратации равна разности энтальпия растворения безводной соли и кристаллогидрата. dH0гидр =dH0раств б/с-dH0раств крист 6) Закон Гесса и его следствия распространяются и на другие функции состояния(S0 dG0 ). dФ0=ΣdФобр пр –ΣdФобр исх. Закон Гесса позволяет обращаться с термохимическими уравнениями как с алгебраическими. Энергия связи – энергия связанной системы каких-либо частиц (например, атома), равная работе, которую необходимо затратить, чтобы разложить эту систему на бесконечно удаленные друг от друга и не взаимодействующие между собой составляющие ее частицы.
6) Термодинамический критерий осуществимости процесса. Энергия Гиббса. Энтальпийный и энтропийный факторы и их роль в экзо- и эндотеримческих реакциях.|
7) Энергия Гиббса как критерий осуществимости химических реакций. Анализ уравнения Гиббса для экзо- и эндотермических реакций при ΔS > 0 и ΔS < 0. |
8)Факторы, определяющие направление протекания химических реакций, анализ уравнения Гиббса для замкнутой системы. Изотерма Вант-Гоффа. |
Энергия Гиббса – изобарно-изотермический потенциал. dG=dH-TdS. dG характеризует возможность самопроизвольного протекания процесса.dG<0 – протекает, >0, самопроизвольно не протекает. В случае экзотермических реакций при dS<0 следует понижать T. Для эндотермических реакций, осуществимых за счёт энтропийного фактора и только при dS>0 необходимы T>1000К. При комнатной температуре обычно TdS невелико и изменение энтальпии больше, нежели энтропии. Поэтому большинство реакций, протекающих при комнатной Т, экзотермичны. dH образования и реакции составляют 80-800 кДж/моль(редко больше 4000 и меньше 40). dH сгорания всегда <0 и составляет тысячи кДж/моль. Энтальпия фазовых превращений обычно < энтальпии реакций. Парообразования – несколько десятков, кристаллизации и плавления ~5-25 кДж/моль. dHT=dH0+dCp*dT, где dCp – изменение теплоёмкости системы. Если фазового перехода не произошло, то dCp=0. Энтропия индивидуальных веществ всегда>0.(от десятков до сотен кДж/моль). Наибольшие значения dS имеют фазовые переходы. Если реакция экзотермична, то знак dG определяет энтальпийный фактор. Также при низких Т вклад энтропийного фактора мал и о направлении реакции можно судить по dH. Эндотермические реакции идут тем успешнее, чем выше Т и если происходит фазовый переход (тогда TdS относительно велико). Для оценки осуществимости процесса используют, однако, не dG, а dG0. Однако эта величина не может использоваться как критерий осуществимости процесса с большим возрастанием энтропии(Напр. Фазовые переходы и реакции термического разложения). Такие реакции могут быть осуществлены при условии, что Т>(dH/dS). Однако стоит заметить, что значения dG0>>0 и dG0<<0 говорят нам о принципиальной возможности протекания реакции. Обычно порогом реакции считают dG0>25 кДж/моль (до этого значения процесс можно осуществить). Изотерма Вант-Гоффа(для 8 вопроса). Чтобы определить dG в нестандартных условиях используют уравнение зависимости dG от концентрации и температуры. (Нест. Условия –это, например, концентрация >1 моль и T не 298К, что повышает/понижает реакционную способность в-ва). Тогда dGi=dG0i+RT*ln(ai), где ai-активная концентрация в-ва. Для перехода из первого состояния во второе можно записать dG2-dG1=dG1-2=dG01-2+RT*ln(a1/a2). Для произвольной химической реакции: aA+bB=mM+nN записывается: dG=dG0+RT*ln(aMm*aNn/aAa*aBb) (т.к. согласно закону Гесса dG0=ΣdGобр пр –ΣdGобр исх). Если концентрации равновесные(dG=0), то dG0=-RT*ln(aMm*aNn/aAa*aBb). Выражение поз логарифмом называется константа равновесия К. dG0=-RT*ln(К). Окончательно, dG=-RT*lnK+ RT*ln(aMm*aNn/aAa*aBb). Если реакция проходит в гальваническом элементе, то dG=-Amax=-nFE (n-число моль-электронов, переходящих от окислителя к восст-лю, F-число Фарадея, Е-разность потенциалов [В]).
II Кинетика
1) Понятие скорости химических реакций. Зависимость скорости химических реакции от концентраций реагирующих веществ в гомогенных системах. Кинетическое уравнение. Константа скорости химической реакции.
2) Понятие скорости химических реакций. Порядок и молекулярность химических реакций.
Кинетика отвечает на вопросы: какие факторы влияют на скорость реакции, каков путь реакции, каковы промежуточные продукты? Она изучает скорость ХР в зависимости от концентрации реагирующих в-в, влияния катализаторов и других факторов, механизмы их протекания. Скорость реакции определяется по изменению концентраций реагирующих в-в во времени. Скорость реакции aA+bB=dD определяется по соотношению vi=+-(dci/n*dt)(производная концентрации по времени). Влияние концентрации на скорость реакции. Для обратимой гомогенной реакции скорость прямой и обратной реакции равна. Для H2+I2=2HI: v=k[H2]*[I2]=k[HI]^2 (Закон действующих масс), в скобках стоят мгновенные молярные концентрации. K-константа скорости реакции, зависящая только от природы реагентов и температуры. В случае гетерогенной реакции(реакции в-в в нескольких агрегатных состояниях) в уравнение ЗДМ входит лишь концентрация газообразных в-в. Обычно реакция протекает через многостадийные процессы. В таких случаях пользуются кинетическим уравнением: v=+-1/n’(dci/dt)=kicin, где n’- стехиометрический коэффициент, n – порядок реакции. Формальным порядком реакции называется показатель степени в кинетическом уравнении реакции, определяемый экспериментально. Прологарифмировав это уравнение получаем линейную форму lgv=lgk+n*lgc, где n=tgφ. Также порядок реакции может быть определён следующим способом – интегрированием кинетического уравнения. -dc/dt=kcn. Тогда мы получаем зависимость с от времени. n=1: ln(c0/ct)=kt; n=2: 1/ct-1/c0=kt; n=3: 1/ct2-1/c02=kt. Тогда, если построить графики lnc,1/c,1/c2 от t, они будут иметь вид прямой. Метод определения порядка реакции – метод избытка реагента. aA+bB=dD. Сначала избыток в-ва А, потом B, в итоге получаем, что v=K*cAm*cBn. Порядок реакции равен m+n. Молекулярность-число молекул, принимающих участиве в элементарном акте химического превращения. Бывает 1,2 и 3-молекулярные. В реакции молекулярность – сумма стехиометрических коэффициентов. Она часто не совпадает с порядком реакции, т.к. стех. Ур-ние не отражает механизма реакции. Типы ХР. 1)Параллельные(в-ва одновременно реагируют в нескольких направлениях). V=k1[A]+k2[B]. Скорость реакции определяется самой быстрой реакцией. V1/V2=k1/k2=const (Принцип независимости ХР).2)Последовательные(реакции с промежуточными стадиями). Константа скорости и скорость посл. Реакций определяется самой медленной реакцией. 3)Цепные(радикальные реакции, в кот. Превращение исходных в-в в продукты осуществляется путём многократного чередования свободных радикалов или атомов) Скорость реакции резко возрастает. 4)Фотохимические(под действием света, каждый квант вызывает превращение одной молекулы(закон Эйнштейна), количество продуктов пропорционально интенсивности падающего света и времени его воздействия(Закон Гершеля и Дрепера). 2 вида фхр: где свет – возбудитель реакции, и где свет – условее протекания реакции.
3) Влияние температуры на скорость химических реакций. Уравнение Аррениуса, физический смысл входящих в него величин. Энергия активации.
4) Влияние температуры на скорость химических реакций. Уравнение Аррениуса. Понятие об энергии активации. Катализ, принцип действия катализаторов.
Скорость ХР сложным образом зависит от температуры. Строгим и физически обоснованным является зависимость k от T в виде уравнения Аррениуса : k=A*exp(-Ea/RT), где А – предэкспоненциальный множитель или фактор частоты, R – универсальная газовая постоянная, Eа-энергия активации, Т – абсолютная температура(К). Энергия, которую надо сообщить молю в-ва, чтобы все его молекулы стали активными в данной реакции называют энергией активации(Ea). Чем больше ЭА, тем медленнее протекает реакция. Если 50<Ea<100 (кДж/моль), то при изменении температуры на 10 градусов, скорость изменится в 2-4 раза(Вант-Гофф). Если известны 2 способа протекания одной реакции, то Еа=(R*T1*T2*ln(k1/k2)/T2-T1). Предэкспоненциальный множитель А характеризует общее число соударений. Для простых реакция А=Z, для реакций со сложными в-вами A=Z*P, Р(стерический фактор, принимает значения от 1 до 10-9) учитывает то, что для взаимодействия необходимо правильное взаимное расположение молекул. Тогда уравнение Аррениуса принимает вид: k=ZP*exp(-Ea/RT)(*). Размерность предэкспоненты та же, что и у константы скорости. K=v/cn. Из (*) видно, что есть 2 способа ускорения реакции – увеличение Т и уменьшение Ea. Последнее осуществляется с помощью катализаторов. Само явление – катализ. Различают гомогенный и гетерогенный катализ (в зависимости от фазы катализатора). Обычная реакция: A+B->A-B(активированный комплекс)->AB. С катализатором: A+K->A-K->AK; AK+B->K-A-B->AB+K. Катализатор не тратится. Увеличение скорости: vk/v=(exp(Ea-Ea.K.)/RT). */ На пути реакции A+B=D лежит энергетический барьер, который необходимо перейти, чтобы произошла реакция(Активированный комплекс)./*
5) Химическое равновесие и его константа. Факторы, влияющие на смещение химического равновесия. Принцип Ле-Шателье.
Чаще всего приходится иметь дело с обратимыми реакциями. aA+bB=cC+dD. Для прямой и обратной реакций: vпр=kпр[A]a[B]b, vобр=kобр[C]c[D]d.(в квадратных скобках – мгновенные концентрации). В состоянии равновесия vпр=vобр. Константа равновесия К=kпр/kобр= [C]c[D]d/[A]a[B]b. При заданной Т К будет постоянной. dG0=-RT*lnK. К выражается и через парциальные давления(Kp). N2+3H2=2NH3: Kp=P2NH3/(PN2*P3H2), Kc=(cNH3)^2/(cN2*c3H2). Kc=Kp*(RT)dn, где dn – изменение числа молей в реакции. Принцип Ле-Шателье. Если на систему оказывать внешнее воздействие, то в системе возникают процессы, уменьшающие внешнее воздействие. Изменение Т. К=kпр/kобр= A*exp(-(Ea.пр-Еа.обр)/RT)=А*exp(-dH/RT). Т.е. чем больше dH, тем больше влияние Т. Охлаждение благоприятствует экзотермическому процессу. Нагревание – эндотермическому. Изменение Р. Смещает равновесие реакций, если dn!=0. Повышение Р смещает равновесие в сторону процесса, при котором уменьшается V. Изменение С. Избыток исходного в-ва смещает равновесие влево, продукта – вправо. При этом К=const(т.к. зависит от Т). Введение катализатора. Не вызывает смещение равновесия(т.к. катализатор ускоряет реакцию).
Химические источники тока
1) Устройство, принцип действия и схема обозначений типичного химического источника электрической энергии (на примере гальванического элемента Даниэля-Якоби). Каков механизм появления двойного электрического слоя и электродного потенциала? |
2) Гальванический элемент Даниэля-Якоби: схема, процессы, происходящие на электродах, суммарный электрохимический процесс. Уравнение Нернста для
каждого электрода и для гальванического элемента в целом. Роль солевого мостика в гальваническом элементе. |
3) Стандартный электродный потенциал. Зависимость электродного потенциала от концентрации потенциалопределяющих ионов и температуры. Уравнение Нернста для отдельного электрода в нестандартных условиях. |
4) Стандартный электродный потенциал, измерение стандартного электродного потенциала. Устройство стандартного водородного электрода. Составьте схему для определения стандартного электродного потенциала медного электрода. |
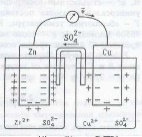 В
основе электрохимии лежат ОВР, т.е. ХР,
сопровождающиеся переходом электронов
от одного атома(восстановителя) к
другому(окислителю). ХИТ – устройства,
получающие электроэнергию, используя
ОВР. Устройство
ГЭ Даниэля-Якоби.
2 сосуда, в которых электрод, помещённый
в раствор электролита, соединёны солевым
мостиком (или пористой перегородкой).
Солевой мостик – стеклянная трубка,
заполненная р-ром электролита, катионы
и анионы которого одинаково подвижны(обычно
KCl).
Соединив электроды проводником первого
рода, начнётся ОВР, о чём говорит
возникновение ЭДС. В основе ГЭ Д-Я
реакция: CuSO4+Zn<->Cu+ZnSO4.
Запиисывается ГЭ так: (-)Zn|Zn2+||Cu2+|Cu(+).
Слева записывается анод(электрод,
на кот. Проходит процесс окисления(отдача
е)), справа – катод(электрод,
на кот. Проходит процесс восстановление).
Направление тока от анода к катоду.
Реакции на
электродах.
Электродные реакции проходят на границе
раздела «электрод-металл». На аноде:
Znтв-2e=Zn2+р,
вследствие чего раствор обогащается
катионами Zn2+.
На Катоде: Cu2+p+2e=Cuтв,
в следствие чего р-р обогащается анионами
SO42-.
Тогда к аноду движутся аноды, а к катоду
– катионы. Электродный
потенциал.
В системе Ме-электролит возможен переход
катионов металла в раствор или из
раствора на поверхность металла(в
зависимости от природы Ме и электролита).
Эти процессы определяются соотношением
энтальпии отрыва иона от крист. Решетки
(dHреш)
и энтальпии сольватации этого иона
Mn+aq(dHсольв).
На границе фаз Ме-электролит устанавливается
равновесие: M+aq
<->ne+Mn+*aq.
Если С ионов Ме в р-ре меньше равновесной,
то возникает отрицательный заряд на
границе Ме по отношению к р-ру(и Ме ->
в раствор). Если С > равновесной, то
происходит переход ионов из р-ра на Ме,
заряжженный положительно(относительно
р-ра). Вследствие этого на границе двух
сред возникает электродный(гальвани-)
потенциал(т.е. двойной электрический
слой на границе двух фаз). Уравнение
Нернста. (для металлического)Еокисл.ф./восст.ф.=Е0
ок.ф./в.ф.+(0,059/n)lg(Amen+).
Получение оного: Для произвольной
химической реакции: aA+bB=mM+nN
записывается: dG=dG0+RT*ln(aMm*aNn/aAa*aBb),
причём dG=-Amax=-qE=-neFE,
т.е.: - neFE=
-neFE0+RT*ln(aMm*aNn/aAa*aBb),
отсюда E=E0-(RT/nF)ln(aMm*aNn/aAa*aBb)=E0+(0,059/n)*ln
(aAa*aBb
/ aMm*aNn).
Причём активность твёрдых в-в = 1.
Уравнение
Нернста для отдельного электрода
условились писать для процесса
восстановления(+ne);Роль
солевого мостика в ГЭ. Поддержание
баланса отрицательных и положительных
ионов в растворе. Стандартный
электродный потенциал.
В
основе электрохимии лежат ОВР, т.е. ХР,
сопровождающиеся переходом электронов
от одного атома(восстановителя) к
другому(окислителю). ХИТ – устройства,
получающие электроэнергию, используя
ОВР. Устройство
ГЭ Даниэля-Якоби.
2 сосуда, в которых электрод, помещённый
в раствор электролита, соединёны солевым
мостиком (или пористой перегородкой).
Солевой мостик – стеклянная трубка,
заполненная р-ром электролита, катионы
и анионы которого одинаково подвижны(обычно
KCl).
Соединив электроды проводником первого
рода, начнётся ОВР, о чём говорит
возникновение ЭДС. В основе ГЭ Д-Я
реакция: CuSO4+Zn<->Cu+ZnSO4.
Запиисывается ГЭ так: (-)Zn|Zn2+||Cu2+|Cu(+).
Слева записывается анод(электрод,
на кот. Проходит процесс окисления(отдача
е)), справа – катод(электрод,
на кот. Проходит процесс восстановление).
Направление тока от анода к катоду.
Реакции на
электродах.
Электродные реакции проходят на границе
раздела «электрод-металл». На аноде:
Znтв-2e=Zn2+р,
вследствие чего раствор обогащается
катионами Zn2+.
На Катоде: Cu2+p+2e=Cuтв,
в следствие чего р-р обогащается анионами
SO42-.
Тогда к аноду движутся аноды, а к катоду
– катионы. Электродный
потенциал.
В системе Ме-электролит возможен переход
катионов металла в раствор или из
раствора на поверхность металла(в
зависимости от природы Ме и электролита).
Эти процессы определяются соотношением
энтальпии отрыва иона от крист. Решетки
(dHреш)
и энтальпии сольватации этого иона
Mn+aq(dHсольв).
На границе фаз Ме-электролит устанавливается
равновесие: M+aq
<->ne+Mn+*aq.
Если С ионов Ме в р-ре меньше равновесной,
то возникает отрицательный заряд на
границе Ме по отношению к р-ру(и Ме ->
в раствор). Если С > равновесной, то
происходит переход ионов из р-ра на Ме,
заряжженный положительно(относительно
р-ра). Вследствие этого на границе двух
сред возникает электродный(гальвани-)
потенциал(т.е. двойной электрический
слой на границе двух фаз). Уравнение
Нернста. (для металлического)Еокисл.ф./восст.ф.=Е0
ок.ф./в.ф.+(0,059/n)lg(Amen+).
Получение оного: Для произвольной
химической реакции: aA+bB=mM+nN
записывается: dG=dG0+RT*ln(aMm*aNn/aAa*aBb),
причём dG=-Amax=-qE=-neFE,
т.е.: - neFE=
-neFE0+RT*ln(aMm*aNn/aAa*aBb),
отсюда E=E0-(RT/nF)ln(aMm*aNn/aAa*aBb)=E0+(0,059/n)*ln
(aAa*aBb
/ aMm*aNn).
Причём активность твёрдых в-в = 1.
Уравнение
Нернста для отдельного электрода
условились писать для процесса
восстановления(+ne);Роль
солевого мостика в ГЭ. Поддержание
баланса отрицательных и положительных
ионов в растворе. Стандартный
электродный потенциал.
![]() (vi-стех.
Коэффициент) Абсолютное значение
потенциала на границе раздела двух сред
измерить нельзя, но можно измерить
разность электродных потенциалов
элементов, то есть ЭДС, возникающую в
контуре. Если же ЭП одного из электродов
выбрать эталонным и принять его Е за 0,
то СЭП будет равен ЭДС контура. Такой
эталонный электрод – водородный
электрод. Устройство:
платиновый электрод, покрытый платиновой
чернью(мелкодисперсной платиной),
погруженный в р-р серной кислоты с а=1
моль/л, обдувается струёй газообразного
Н под давлением 1 атм=1,03*105Па.
Реакция: Н2<->
2Н++2e.
Схема определения СЭП: (-)Pt,
H2|2H+||Mn+|M(+)
Если СЭП<0, то металл записывается
слева( и электрод с металлом – анод).
(vi-стех.
Коэффициент) Абсолютное значение
потенциала на границе раздела двух сред
измерить нельзя, но можно измерить
разность электродных потенциалов
элементов, то есть ЭДС, возникающую в
контуре. Если же ЭП одного из электродов
выбрать эталонным и принять его Е за 0,
то СЭП будет равен ЭДС контура. Такой
эталонный электрод – водородный
электрод. Устройство:
платиновый электрод, покрытый платиновой
чернью(мелкодисперсной платиной),
погруженный в р-р серной кислоты с а=1
моль/л, обдувается струёй газообразного
Н под давлением 1 атм=1,03*105Па.
Реакция: Н2<->
2Н++2e.
Схема определения СЭП: (-)Pt,
H2|2H+||Mn+|M(+)
Если СЭП<0, то металл записывается
слева( и электрод с металлом – анод).
5) Таблица стандартных электродных потенциалов для окислительно-восстановительных систем. Металлический электрод, уравнение Нернста для металлического электрода. |
Металлический электрод – металл, погруженный в раствор своей соли M|Mn+, например, цинковый или медный. Еокисл.ф./восст.ф.=Е0 ок.ф./в.ф.+(0,059/n)lg(Amen+)
6) Электроды 1-го и 2-го рода на примере медного и хлорсеребряного электродов. Уравнение Нернста для них. Почему электроды 2-го рода можно использовать в качестве электродов сравнения? |
7) Электроды сравнения: водородный, хлорсеребряный, стеклянный. Устройство, уравнение Нернста. Почему электроды 2-го рода можно использовать в качестве электродов сравнения? |
8) Стеклянный электрод: устройство, уравнение Нернста. |
Электроды
1-го рода: 1)
электроды, состоящие из элементарного
в-ва, находящегося в контакте с раствором,
содержащим его собственные ионы
(например, металлический(медный.
Cu2+p+2e=Cuтв,
в следствие чего р-р обогащается анионами
SO42-,
а на поверхности электрода оседает
металлическая медь. Еокисл.ф./восст.ф.=Е0
ок.ф./в.ф.+(0,059/n)lg(Amen+))
или газовый). 2) Редокс-электроды. Состоят
из инертного по отношению к данному
электролиту проводника первого
рода(платина, графит), погруженного в
раствор, в котором находятся одновременно
окисленная и восстановленная формы
потенциалопределяющего в-ва. Пример:
Pt|Sn4+,Sn2+;
Pt|Fe3+,Fe2+.
Уравнение Нернста: Eок.ф./вост.ф.=E0.ок.ф./вост.ф.
+(0,059/dn)lg(aок.ф./авосст.ф).
Окисленная
форма – та, где заряд больше.
Электроды 2-го
рода. Металлические
пластины, покрытые слоем труднорастворимой
соли того же металла и опущенные в
раствор, содержащий анион этой соли.
Его потенциал определяется активностью
аниона в р-ре. Хлорсеребряный
электрод. Ag|AgCl|Cl-.
(серебряный проводник покрыт слоем
AgCl,
который погружен в насыщенный р-р KCl.
Ag++e=Ag0.
EAg+/Ag=E0.Ag+/Ag+0,059*lg(aAg+).
Но т.к.
активность связана с произведением
растворимости(ПР) хлорида
серебра(ПРAgCl=aAg+*aCl-),
то EAg+/Ag=E0.Ag+/Ag+0,059*lg(ПРAgCl)-0,059*lg(aCl-).
=>Eхсэ
=Eхсэ-0,059*lg(aCl-).
Здесь ионы хлора потенциалопределяющие.
AgClтв+е=Ag++Cl-.
Таким образом, Электроды 2-го рода можно
использовать в кач-ве электродов
сравнения, т.к. они обратимы по отношению
к аниону, активность которого можно
поддерживать постоянной, что приводит
к постоянству электродного потенциала.
Стеклянный
электрод. Стеклянный
электрод – электрод для определения
концентрации H+
(pH)
относится к ионоселективным(мембранным)
электродам. Он состоит из стеклянного
тонкостенного шарика(мембраны),
припаянного к стеклянной трубке. В
шарике – р-р HCl
(aH+=0,1
моль/л), в который опущен хлорсеребряный
электрод. При погружении электрода в
раствор с измеряемой CH+
между мембраной и исследуемым в-вом
возникает разность потенциалов(из-за
процесса ионного обмена).
Ест=Е0.ст.+0.059*lg[H+]=Е0.ст.-0.059*pHвнеш.
Водородный
электрод. Устройство:
платиновый электрод, покрытый платиновой
чернью(мелкодисперсной платиной),
погруженный в р-р серной кислоты с а=1
моль/л, обдувается струёй газообразного
Н под давлением 1 атм=1,03*105Па.
Реакция: Н2<->
2Н++2e.
Схема определения СЭП: (-)Pt,
H2|2H+||Mn+|M(+)
Если СЭП<0, то металл записывается
слева( и электрод с металлом – анод).
Дополнение.
Концентрационный элемент – одинаковые
электроды с разными активностями
потенциалопределяющего иона. Существуют
такще сухие элементы (батарейки)
 .
.
9) Аккумуляторы (на примере свинцового аккумулятора). Реакции на аноде и катоде, суммарная реакция в свинцовом аккумуляторе при его работе (разрядке) и зарядке. ЭДС свинцового аккумулятора (конечная формула с использованием активности серной кислоты и воды). |
Аккумулятор – обратимый гальванический элемент многоразового действия. При пропускании через них электрического тока(зарядке) аккумуляторы накапливают хим. Энергию, которую потом при работе(разрядке) отдают в виде электрической энергии. Наиболее распространённые - свинцовый(кислотный) и щелочной. Свинцовый аккумулятор. Активная масса анода – перфорированные свинцовые пластины, катода – диоксид свинца, впрессованный в свинцовые решётки. Электролит – раствор (35%) H2SO4. Схема: (-)Pb|PbSO4|H2SO4|PbSO4|PbO2|Pb(+).При работе протекает реакции: (-)Pb+SO42--2e=PbSO4. E0.PbSO4/Pb=-0,36B; (+) PbO2+4H++SO42-+2e=PbSO4+2H2O. E0.PbO2/PbSO4=1,69B. Суммарная реакция: Pbтв+PbO2.тв+2H2SO4 2PbSO4тв+2H2O. Уравнение Нернста записывается так(учитывая, что атв=1): Е= E0.PbO2/PbSO4-E0.PbSO4/Pb+0,059lg(aH2SO4/aH2O)=2.05+0,059lg(aH2SO4/aH2O) . В процессе работы аккумулятора концентрация кислоты падает, а следовательно, падает и ЭДС. Когда ЭДС достигает 1,85 В, аккумулятор считается разрядившимся. При более низкой ЭДС пластины покрываются тонким слоем PbSO4 и и аккумулятор разряжается необратимо. Во избежание этого аккумулятор периодически подзаряжают. Дополнительно: щелочной железо-никелевый аккумулятор. Электролит – 20% КОН. Активная масса анода – губчатое железо, катода – гидроксид никеля. (-)Fe|Fe(OH)2|KOH|Ni(OH)2|Ni(OH)3|Ni(+). Суммарная реакция: Fe+2Ni(OH)3Fe(OH)2+Ni(OH)2. В уравнении Нернста стоят не активности, а ПР данных нерастворимых в-в. E=E0.Ni(OH)3/Ni(OH)2-EFe(OH)2/Fe +(0.059/2)lg(ПР2Ni(OH)3/(ПР2Ni(OH)2*ПРFe(OH)2). Его преимущество – ЭДС не зависит от концентрации щёлочи.