

Via un conseil génétique :
Analyse du contexte clinique grave ou modéré
Grossesse ? un diagnostic prénatal peut être proposé
Prescription pour rechercher des mutations courantes CFTR ou rares, familiales si connues
Une fois les mutations trouvées :
Une étude familiale est toujours proposée
Vérification des mutations trouvées chez le propositus (atteint) avec une étude parentale (si possible)
étude de ségrégation familiale
En laboratoire de Biologie moléculaire (avec agréments en Dépistage prénatal (DPN)) :
Extraction de l’ADN à partir de sang le plus souvent
Amplification génique de différents exons du gène CFTR par PCR (polymérase chain reaction)
Analyse biomoléculaire d’une trentaine de mutations courantes (couvertures d’environ 85% du gène CFTR
Analyse moléculaire de mutations rares (plus longue)
IV) Techniques moléculaires du gène cftr :
Techniques moléculaire mutations courantes :
Ligation d’oligonucléotiques (OLA) analyse des fragments
Hybridation reverse
Amplification spécifique d’allèles (ARMS) gel d’agarose
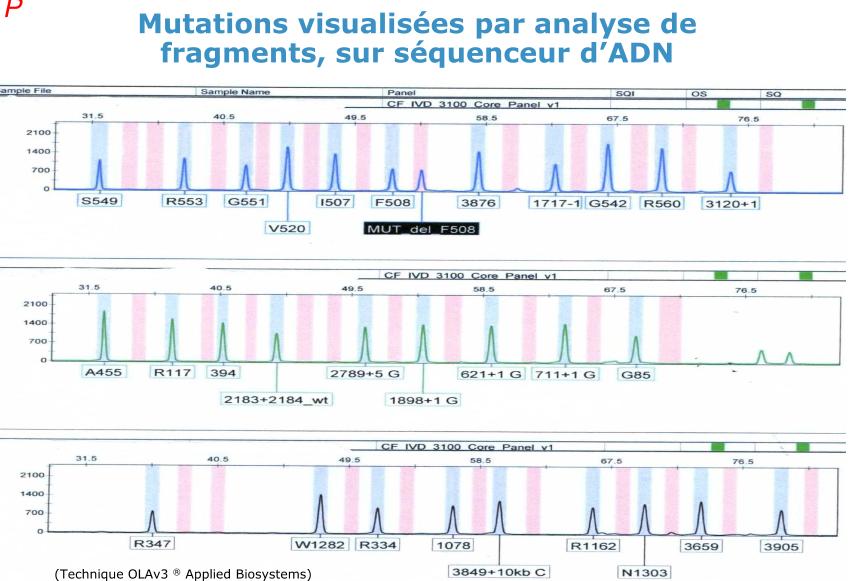
Chaque pic dans une zone grise correspond à l’intervention normale du gène. Si il y a une mutation, on a par exemple le F508 normale et en rose uniquement le F508 délété
Si on a 30 fragments (30 pics) on a 30 PCR de 30 zones à risque tout de suite balayées par le séquenceur qui reconnaît l’allèle normale ou muté.
Techniques moléculaires mutations rares :
DHPLC
DGGE (électrophorèse en gradient d’agents dénaturant)
Recherche de remaniements par PCR multiplex fluorescente
Puces (micro-arrays)
…
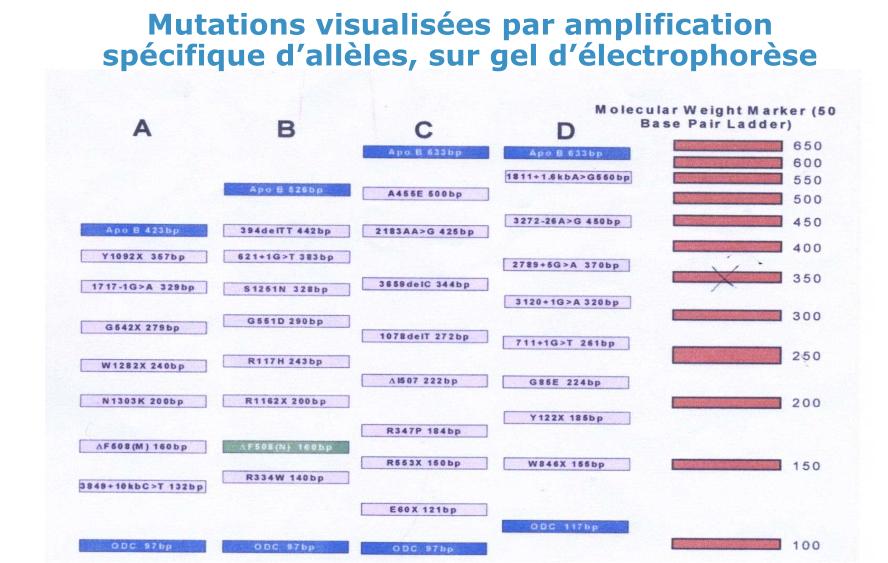
On a 4 pics, 4 colonnes, les bandes s’allument en fonction de l’allèle normale ou anormal
Cela se fait sur électrophorèse, on le fait pour le dépistage néonatal.
Nature des mutations cf
Mutation non sens :
G 542X (codon stop) : origine phénicienne
W1282 X : origine ashkénaze
Mutations faux sens :
N1303K : origine celte
G551D : origine ibère
Mutation d’épissage :
1717-1G > A
Délétion : F508L
La plus fréquente dans le monde, avec un gradient décroissant entre le Nord (87%) et le Sud (Turquie 30%)
Origine caucasienne
Insertion : 435 insA
Del ou ins décalage de la phase de lecture (POL ou ORF-Open Reading Frame-)
Grands réarrangements…

Il y a des bornes 5’ et en 3’, si l’excision est bien faite , on a un raboutage entre les exons Si il y a une mutation sur un site d’épissage , on a des raccords entre le mauvais exons , on peut même perdre un exon . Sur le 1717e nucléotide, -1 après le G devient A, sur le 1811e nucléotides, +1 le G devient C
Fréquence des mutations CF (vraiment intéressant..)
G542X : environ 3% (1 à 6,6%)
N1303K : environ 2% (0,75% à 4,5%)
1717-1G>A : environ 1,5% (0 à 2,7%)
2789+5G>A : environ 1% (0 à 2,8%)
9 autres mutations ont une fréquence > 0,4% :
G551D, W1282X, R553X, I507del, 2183 AA>G, 1078 delT, 711+1G>T, R1162X, Y1092X
Classification des mutations par régions CFTR
NBF 1 :
Exon 10 : F508 del
Exon 11 : G542X, R553X, G551D, 1717-1G>A
exons 10/11 représente 75% des mutations en France
NBF 2
Exon 19 : R1162X
Exon 20 : W1282X
Exon 21 : N1303K
TM 1 :
Exon 4 : R117H, 621+1G>T
TM 2 :
Intron 14b : 2789+5G>A
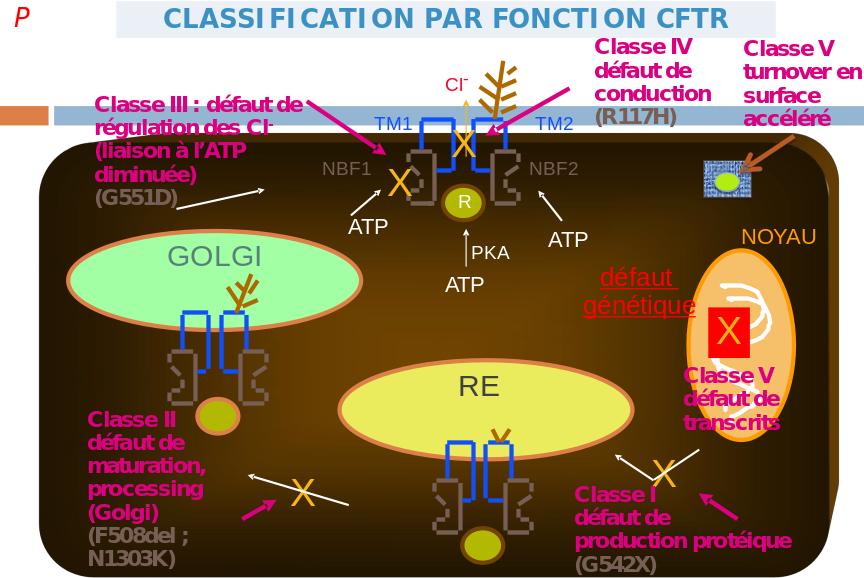
Si on a un défaut majeur, on n’aura jamais de production protéique suffisante. La protéine est morte née et les chlorures ne sortent pas de la membrane plasmique
Classe I : défaut de production protéique avec un codon stop => pas de protéine ou protéine tronquée, elle est détruite
Classe II : défaut de maturation ; les protéines sont détruites aussi dans les lysosomes
Classe III : défaut de régulation, de conduction ; régulation des chlorure très limités, l’ATP a du mal à accroché le NBF
Classe IV : défaut de conduction, la protéines a réussi à être synthétisée, elle a été envoyée dans la membrane plasmique mais le canal ne marche pas bien. Cependant il y a suffisamment de chlorure pour vivre.
Il faut un minimum de protéines correctes pour pouvoir vivre d’ou la thérapie génique (cf. après)
Répartition française des mutations et génotypes :
5 génotypes environ 58% des cas CF en France :
F508del / F508del : 47,8%
F508del / G542X : 3,4%
F508del / N1303K : 2,7%
F508del / 1717-1G>A : 2,1%
F508del / 2789+5G>A : 1,5%
Corrélations des génotypes-phénotypes :
Mutations S (sévères) et M (modérées)
Génotype S/S = phénotype S
Génotype M/M = phénotype M
Génotype S/M = phénotype M
Ces corrélations génotypes/phénotypes :
Sont plus évidentes au niveau du statut pancréatique que pulmonaire
Influence donc d’autres facteurs génétiques (CF « modifiers ») et environnementaux
Donc les personnes muco ont besoin d’un environnement très sain : pas de tabac, de contact avec les gens grippaux ou les autres muco (pas de colo entre les muco)
Corrélation génotype-phénotype et transcription :
En fonction des anomalies génétiques, l’évaluation des taux de transcrits CFTR normaux persistants dans les tissus permet de mieux comprendre la diversité des phénotypes.
Il n’y a pas de corrélation absolue génotypes/phénotypes mais il y a une corrélation raisonnable entre le % résiduel de la fonction CFTR normale et l’atteinte des différents organes.
Les taux de transcrits CFTR suivants peuvent correspondre à différentes symptomatologiques :
Les taux de transcrits CFTR suivant corresponde à différentes symptomatologie
Entre 50% et 100% => aucune symptomatologie
10% à 50% => stérilité masculine (CBAVD)
< 5% => test de sueur anormale
3-4% => apparition d’une maladie pulmonaire
<1% => Ajout d’une insuffisance pancréatique
On revient à l’importance de la thérapie génique, si on arrive à rajouter environ 5% de protéine normales, les patients restent en vie. Il faut ajouter un minimum de protéines normales pour restaurer la fonction pancréatique.
On ne peut restaurer des capacités pulmonaires normales à un enfant qui a déjà des poumons en carton. L’espoir de la TG se fait donc chez les jeunes personnes qui n’ont pas les poumons trop abimés