

7.6. Жидкостная хроматография. Методы вэжх, иох и тсх
Знакомство с отдельными методами хроматографического анализа логично начать с жидкостной хроматографии (ЖХ), так как этот метод возник первым.
Сорбенты. В жидкостной хроматографии неподвижную фазу образуют мелкодисперсные частицы твердых сорбентов – оксида алюминия, силикагеля, алюмосиликатов, угля, сажи, мела, талька. Используют также природные (целлюлоза, крахмал) или, чаще, специально синтезированные полимерные сорбционные материалы (тефлон и др.). Размер частиц сорбента не превышает 1 мм, часто используют порошки с еще меньшим размером частиц (десятки микрометров). Для эффективного разделения компонентов смеси надо, чтобы все частицы были приблизительно одинакового размера. Это достигается с помощью многократного просеивания сорбента через сита с разным размером отверстий. Разделение веществ в ЖХ чаще всего происходит по адсорбционному механизму, в этом случае сорбенты называют адсорбентами. Удельная поверхность адсорбентов должна быть не меньше 50 м2/г.
Адсорбция молекул или ионов из раствора идет не на всей поверхности твердой частицы, а только на определенных активных центрах этой поверхности. Так, на поверхности оксида алюминия существуют по меньшей мере три вида активных центров (кислотные, основные и т. п.), адсорбирующих из раствора молекулы разного типа. Примером кислотных центров могут быть группы Si-O-H на поверхности силикагеля. В зависимости от способа приготовления и подготовки адсорбента (например, от способа его промывки реагентами, от температуры и продолжительности прокаливания и т. п.) можно получить адсорбент с преобладанием тех или иных центров, с меньшим или большим числом центров на единице поверхности. Такими способами можно получать адсорбенты с заданными свойствами.
Для разделения малополярных органических молекул преимущественно используют полярные адсорбенты типа силикагеля и оксида алюминия, а в качестве элюентов применяют органические растворители (углеводороды, спирты, эфиры). Для разделения ионов и сильнополярных органических молекул используют совсем другие фазы. В качестве неподвижной фазы берут неполярные вещества (крахмал, целлюлоза и т. п.), подвижной фазой могут быть: вода, водные растворы кислот, солей или щелочей, водно-спиртовые или водно-ацетоновые растворы. Такой вариант анализа называют обращенно-фазовой хроматографией.
Изотермы сорбции. При контакте сорбента с раствором, содержащим растворенное вещество Х, устанавливается равновесие. Его основной характеристикой является изотерма сорбции – полученная при некоторой постоянной температуре (например, при комнатной) зависимость параметра a (количества вещества, сорбированного одним граммом сорбента) от начальной концентрации Х в растворе (С, моль/л). Если поглощение Х идет по адсорбционному механизму, такую зависимость называют изотермой адсорбции. При низких концентрациях Х изотерма адсорбции прямолинейна, затем ее наклон, как правило, уменьшается1. Это объясняется ограниченностью числа активных центров: по мере их заполнения частицами Х дальнейшая адсорбция снижается или полностью прекращается. Угловой коэффициент изотермы адсорбции равен коэффициенту распределения Х между фазами. Его называют коэффициентом Генри и обозначают символом Г. Обычно так же называют коэффициент распределения и при других механизмах сорбции, хотя это не совсем правильно.
Когда в растворе есть несколько разных растворенных веществ (например, X1 и X2 ), их изотермы адсорбции, как правило, различаются по наклону линейного участка (Г1 ≠ Г 2 ). В таких случаях компоненты смеси X1 и X2 могут быть хроматографически разделены. Если же для данной системы сорбент–растворитель вещества X1 и X2 имеют одинаковые изотермы адсорбции, разделение X1 и X2 невозможно.
Для получения хорошо воспроизводимых хроматограмм с отчетливо выраженными симметричными пиками и, соответственно, для успешного разделения иследуемых смесей хроматографисты стремятся работать в области низких концентраций, обеспечивающих постоянные значения коэффициентов распределения (на прямолинейных участках изотерм адсорбции). Поэтому для проведения хроматографического анализа стараются взять как можно меньшую массу и объем пробы. Ограничением является лишь необходимость достоверно зарегистрировать сигнал каждого компонента, выходящего из хроматографической колонки.
Иногда изотермы сорбции имеют выпуклую или вогнутую форму даже в области очень низких концентраций. Нелинейность изотерм приводит к искажению формы пиков на хроматограмме, они становятся несимметричными. Искажение затрудняет определение площади пика (по ней рассчитывают результат анализа). При несимметричной форме хроматографического пика с растянутым влево или вправо контуром больше вероятность наложения пиков разных компонентов пробы друг на друга. Непредсказуемость формы изотерм адсорбции и нередкая зависимость коэффициентов распределения от концентрации Х – существенные недостатки жидкостной адсорбционной хроматографии по сравнению с распределительной (газожидкостной).
Метод ВЭЖХ. Ионная хроматография. Классическая адсорбционная жидкостная хроматография (метод Цвета) до сих пор применяется во многих аналитических лабораториях. В этом случае мелкодисперсный сорбент загружают в колонку высотой 1–2 м, вносят в верхнюю часть колонки пробу и медленно промывают колонку подходящим элюентом. Состав элюента может быть неизменным, но нередко колонку промывают сначала одним элюентом, затем другим и т. д. На выходе из колонки периодически контролируют состав элюата, измеряя показатель преломления (nd) или другое свойство раствора. В отдельные приемники собирают фракции с разными значениями nd. Так можно разделить какой-либо нефтепродукт, выделяя сумму предельных (парафиновых) углеводородов, легкие, средние и тяжелые ароматические углеводороды и т. п.
Классический вариант ЖХ длителен, трудоемок и далеко не всегда обеспечивает полное разделение компонентов. Качество разделения улучшается при уменьшении размера частиц. Поэтому на смену классическому варианту ЖХ в 70-е гг. XX века пришел метод, в котором применяют сорбенты с гораздо меньшим размером частиц (5–10 мкм) и сравнительно небольшими колонками (длина 10–30 см, диаметр порядка 5 мм). В лабораториях используют готовые, серийно выпускаемые колонки. Они могут содержать как обычные сорбенты для адсорбционной хроматографии, так и сорбенты с модифицированной поверхностью. Например, на поверхность сорбента можно заранее нанести тончайший слой вязкой неподвижной жидкой фазы или к поверхности сорбента привить с помощью мощного радиоактивного излучения какие-либо крупные органические молекулы. Оба приема превращают адсорбционный механизм разделения в распределительный, что существенно улучшает качество разделения компонентов пробы. В колонку можно поместить и мелкозернистый катионит или анионит, такие колонки используют в ионообменной хроматографии.
Поскольку гидравлическое сопротивление колонки с мельчайшими частицами сорбента очень велико, для ускорения анализа элюент нагнетают в колонку под большим давлением, в 100–400 раз выше атмосферного. Для проведения анализа потребуется специальный прибор – жидкостной хроматограф (рис. 7.5), снабженный мощным насосом и детектором, непрерывно измеряющим свойства элюата.
Такой вариант анализа получил название: жидкостная хроматография высокого давления (ЖХВД) или высокоэффективная жидкостная хроматография (ВЭЖХ).
Пробу в жидкостной хроматограф вводят с помощью специальных кранов-дозаторов. Соединительные трубки и сами колонки выполняют из прочных, химически инертных материалов. Очень важно обеспечить герметическое соединение разных узлов жидкостного хроматографа, которое должно выдержать высокое давление. Иногда состав элюента меняют непрерывно (градиентное элюирование), смешивая в переменном соотношении 2–3 растворителя по заранее заданной программе. Такой прием позволяет полностью вымывать из колонки все компоненты пробы и получать на одной и той же хроматограмме пики соединений, сильно различающихся по растворимости (например, растворимых только в воде и растворимых только в ацетоне).
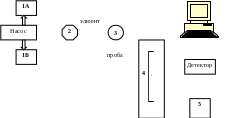
Рис. 7.5. Принципиальная схема хроматографа для ВЭЖХ: 1А и 1Б – резервуары для растворителей; 2 – смеситель для градиентного элюирования; 3 – кран-дозатор; 4 – термостат с колонкой; 5 – устройство для сбора фракций
Жидкостные хроматографы оснащают фотометрическими, флуориметрическими, кондуктометрическими или рефрактометрическими детекторами. Контролируя оптическую плотность элюата или интенсивность люминесценции на определенных длинах волн, можно зафиксировать выход некоторых компонентов пробы и рассчитать их содержание, не фиксируя сигналы других компонентов. Кондуктометрические и особенно рефрактометрические детекторы менее избирательны, с их помощью можно выявить все компоненты пробы. Для записи хроматограмм и их обработки, вплоть до расчета концентрации всех компонентов, в настоящее время используют компьютерную технику.
Приборы для ионообменной хроматографии устроены несколько сложнее, чем другие жидкостные хроматографы. Как правило, детектором в этих приборах служит чувствительный кондуктометр. Кроме узлов, показанных на рис. 7.5, в ионных хроматографах между основной (разделительной) колонкой и детектором установлены дополнительные ионообменные колонки, подавляющие фоновую электропроводность элюата. Так, при анализе смеси катионов металлов разделительную колонку заполняют мелкодисперсным катионитом в Н-форме, а в качестве элюента используют разбавленные растворы НСl. Подавляющую колонку заполняют анионитом в ОН-форме, в ней будут поглощаться Сl–-ионы, а в элюат выделятся ОН‑ионы, которые тут же соединятся с прошедшими через разделительную колонку Н-ионами. В результате перед детектором НСl «преобразуется» в воду, и фоновая электропроводность исчезнет. Напротив, вытесненные из разделительной колонки катионы металлов беспрепятственно пройдут через подавляющую колонку, поочередно пройдут через кондуктометрический детектор, создадут аналитические сигналы, а затем и пики на хроматограмме.
Современные жидкостные хроматографы обеспечивают высокую точность анализа (погрешность обычно не превышает 5 % отн.). Пределы обнаружения компонентов анализируемой смеси зависят от их природы и еще более от типа используемого детектора, они могут составлять 10–9 и даже 10–10 г. Однако столь высокая чувствительность и точность анализа требуются далеко не всегда. Вместе с тем дорогие и сложные жидкостные хроматографы доступны не всем аналитическим лабораториям. Очевидно, кроме метода ВЭЖХ, нужны другие, более простые и дешевые варианты жидкостной хроматографии, которые, тем не менее, позволят быстро разделить исследуемую смесь и опознать ее компоненты. Задача имеет два решения – это методы тонкослойной и бумажной хроматографии.
Тонкослойная хроматография. Метод был предложен в 1938 г. Н.И. Измайловым и М.С. Шрайбер. На плоскую пластинку, поверхность которой покрыта тонким (до 1 мм) слоем твердого мелкозернистого сорбента, наносят каплю разделяемой смеси (≈ 0,01 мл). Наиболее часто в качестве сорбента используют силикагель, оксид алюминия, целлюлозу, в качестве связующего иногда добавляют гипс или крахмал. В наиболее распространенном варианте ТСХ нижний край пластинки погружают в растворитель (элюент). За счет капиллярных сил элюент перемещается вверх по слою сорбента, увлекая за собой растворимые компоненты пробы. Чтобы элюент не испарялся с поверхности сорбента, пластинка на время разделения должна быть помещена в герметически закрытую прозрачную камеру.
Обычно каждую частицу сорбента покрывает очень тонкий слой воды, и компоненты пробы в соответствии с их растворимостью многократно перераспределяются между органической ПФ и водой в неподвижной жидкой фазе (механизм распределительной хроматографии). Однако в ТСХ возможны и другие механизмы (адсорбционный, ионообменный, молекулярно-ситовый, смешанный и т. п.). Многократные акты сорбции и десорбции соответствующих молекул (или ионов) частицами сорбента приводят к тому, что компоненты пробы постепенно отстают от подвижной фазы, поднимающейся по слою сорбента. При правильном выборе ПФ и НФ, а также при достаточно малом объеме пробы каждый компонент движется в виде небольшой зоны («пятна»), и эти пятна не расплываются по поверхности сорбента. Когда фронт элюента приблизится к верхнему краю пластинки, ее вынимают из хроматографической камеры, высушивают и определяют положение пятен на хроматограмме (рис. 7.6). Если компоненты пробы не окрашены, хроматограмму проявляют, облучая УФ-светом, обрабатывая парами иода или другими реагентами.

Рис. 7.6. Схема выполнения (А) и результат (Б) разделения смеси методом ТСХ. На стартовую линию были нанесены: предполагаемые компоненты пробы порознь (1–4) и сама проба (5), содержащая компоненты 1 и 3. Стрелкой показано направление движения элюента
Относительная скорость движения i-го компонента или его подвижность (Rf) равна:
Rf =
![]() , (7.13)
, (7.13)
где
![]() – высота подъема пятна компонента;
– высота подъема пятна компонента;
![]() – высота подъема фронта растворителя
за то же время. Для компонентов,
нерастворимых в ПФ, Rf
= 0, для несорбируемых компонентов
Rf = 1, другие компоненты
пробы дают значения Rf
от 0 до 1.
– высота подъема фронта растворителя
за то же время. Для компонентов,
нерастворимых в ПФ, Rf
= 0, для несорбируемых компонентов
Rf = 1, другие компоненты
пробы дают значения Rf
от 0 до 1.
Различие коэффициентов распределения разных компонентов приводит к неодинаковой подвижности этих компонентов и, следовательно, к их разделению. После проявления будет наблюдаться столько пятен, сколько компонентов с разными коэффициентами распределения изначально присутствовало в пробе.
Выбор растворителя для метода ТСХ определяется природой сорбента и свойствами предполагаемых компонентов пробы. Часто применяют смеси двух-трех растворителей. В частности, при опознании индивидуальных аминокислот в их смеси в качестве ПФ используют смесь уксусной кислоты, воды и н-бутанола.
Если разделения не произошло или нет гарантии, что разделились все компоненты пробы, надо использовать другой элюент. Удобно, в частности, повернуть уже проявленную пластинку на 900 и погрузить ее в новый растворитель. Этот вариант анализа называют двумерной тонкослойной хроматографией (рис. 7.7). Полезно также провести повторное разделение, нанеся новую порцию исследуемой пробы на пластинку с другим сорбентом.

Рис. 7.7. Двумерная хроматография семикомпонентной смеси красителей по методу ТСХ: А — ввод пробы; Б — после обработки первым элюентом; В — после обработки вторым элюентом. Указаны стартовые линии и линии подъема фронта растворителя
Для идентификации компонентов в методе ТСХ пользуются следующими приемами.
1) Сравнение со свидетелями. В этом случае рядом с каплей пробы на стартовую линию наносят капли «свидетелей» – предполагаемых компонентов пробы (химически чистые индивидуальные вещества или их растворы в ПФ). Последующее совпадение по высоте подъема (а также по окраске) какого-либо пятна пробы и пятна свидетеля указывает на возможное присутствие соответствующего вещества в пробе, хотя и не гарантирует этого (разные вещества могут иметь совпадающие окраски и совпадающие высоты подъема). Для подтверждения проверку проводят повторно, в новых условиях – с другими сорбентами, с другим растворителем. Случайное совпадение характеристик свидетеля и компонента пробы в нескольких системах маловероятно.
2) Сравнение с табличными данными. Величина Rf не зависит от размера пластинки, времени разделения и (при достаточно малой массе пробы) от концентрации компонента в пробе и присутствия других компонентов. Таким образом, Rf – это идентификационная характеристика. Для часто применяемых систем ПФ–НФ подвижности индивидуальных веществ известны и собраны в таблицы. Сравнение с табличными значениями Rf – удобный и быстрый метод идентификации, особенно если в лаборатории отсутствует нужный свидетель или вообще не ясно, какие свидетели использовать, поскольку неизвестен даже ориентировочный состав пробы. К сожалению, экспериментальные значения Rf недостаточно воспроизводимы, так как на Rf влияют толщина слоя, влажность и зернистость сорбента, а эти факторы при самостоятельном изготовлении пластинки трудно стандартизовать. Стандартные пластинки с тонким слоем сорбента, нанесенным на металлическую фольгу, выпускаются промышленностью и широко используются в лабораторной практике. Но даже при использовании таких пластинок идентификация веществ по табличным значениям Rf менее надежна, чем идентификация с одновременным нанесением пробы и свидетеля на одну и ту же пластинку.
3) Обработка селективными реагентами. Окраска или свечение пятен могут возникать или меняться при обработке хроматограммы реагентами. Так, некоторые углеводы и аминокислоты имеют совпадающие значения Rf , но при опрыскивании хроматограммы нингидрином пятна аминокислот синеют, а углеводы с этим веществом не реагируют.
4) Спектрофотометрическая идентификация. Сорбент в соответствующей зоне аккуратно снимают с пластинки и переносят в сосуд, где обрабатывают растворителем. Затем раствор с извлеченным компонентом отфильтровывают и регистрируют его спектр, например, спектр поглощения в УФ-области против чистого растворителя. По спектру судят о химической природе компонента. В отдельных случаях регистрируют спектр отражения или спектр флуоресценции самого пятна. Эти же приемы используют и для количественного анализа.
Бумажная хроматография. Вместо пластинок с нанесенным тонким слоем сорбента можно использовать полоски или листы специальной бумаги (типа фильтровальной). Остальные операции в методе бумажной хроматографии проводятся примерно так же, как и в тонкослойной. Для разделения на бумаге веществ, растворимых в воде (например, неорганических ионов), в качестве ПФ берут органический растворитель, а бумагу заранее смачивают водой. Для разделения компонентов, хорошо растворимых в органических растворителях, бумагу заранее пропитывают подходящим органическим растворителем, а в качестве подвижной фазы берут воду, водный раствор какой-либо кислоты или щелочи. Еще лучше использовать буферный раствор с известной величиной рН, так как при разделении веществ, способных участвовать в процессах протолиза, величина рН водной фазы сильно влияет на величину Rf .
Для этого варианта хроматографии специфическими приемами являются:
1) сочетание хроматографии с электрофорезом. Для этого во время хроматографического разделения компонентов пробы к влажному листу фильтровальной бумаги прикладывают постоянное электрическое напряжение. Дополнительное воздействие электрического поля приводит к более четкому разделению, особенно четко делятся ионы разного заряда. Электрофорез можно проводить не только одновременно с хроматографическим разделением пробы, но и последовательно (до или после хроматографирования);
2) круговая хроматография с испарением растворителя. В этом случае на горизонтально лежащий лист бумаги наносят каплю анализируемого раствора, высушивают ее, а затем медленно и непрерывно подают в центр полученного пятна элюент. Под действием капиллярных сил он движется в радиальном направлении от центра пятна и увлекает за собой компоненты смеси. Через небольшое время на хроматограмме получаются концентрические кольца, число которых равно числу компонентов, отличающихся по своей подвижности.
Отметим, что метод бумажной хроматографии, изобретенный в 1941 г. Мартином и Синджем, в настоящее время используют в аналитических лабораториях довольно редко, но он хорошо подходит для первого знакомства с хроматографическими методами, например, при изучении аналитической химии школьниками или студентами.