

6.3.5. Люминесцентный анализ*
Принцип метода и области его применения. В определенных условиях часть поглощенной веществом энергии может выделиться в виде вторичного излучения. Это явление называют люминесценцией. Кванты вторичного излучения, испускаемого люминесцирующими атомами, молекулами или ионами, имеют меньшую энергию, чем кванты, которые те же частицы поглощали при своем возбуждении. Виды люминесценции классифицируют по способу возбуждения. Наиболее известны:
1) катодолюминесценция (свечение под действием потока электронов, например, свечение экрана кинескопа);
2) хемилюминесценция (свечение в результате протекания некоторой химической или биохимической реакции, например у светлячков);
3) фотолюминесценция. В этом случае проба светится за счет облучения невидимым УФ-светом от внешнего источника. Именно этот вид люминесценции обычно применяют в химическом анализе.
Виды люминесценции классифицируют и по времени жизни возбужденного состояния. В анализе преимущественно используют флуоресценцию – в этом случае возбужденное состояние молекулы неустойчиво, и вторичное излучение пробы в УФ- или видимой области спектра прекращается сразу после удаления источника возбуждения. Иногда используют и фосфоресценцию – в этом случае свечение вещества продолжается в течение нескольких секунд, минут или даже часов после прекращения возбуждения.
Явление люминесценции известно с давних времен, но физики стали изучать его лишь во второй половине XIX века. В 1864 г. Дж. Стокс (Англия) установил связь интенсивности свечения с концентрацией флуоресцирующих веществ в растворе и предложил использовать такую связь в аналитических целях, подчеркнув высокую чувствительность нового метода. В настоящее время в качестве аналитического сигнала используют интенсивность вторичного излучения на некоторой длине волны.
Первые методики люминесцентного анализа были созданы в 30-х гг. XX века, во многом благодаря работам академика С.И. Вавилова и его учеников. Сегодня этот метод используют не очень широко, но в некоторых областях он просто незаменим. По спектрам люминесценции опознают и определяют особо опасные органические вещества в объектах окружающей среды (на уровне 10–6 % и ниже). В частности, так находят содержание полициклических ароматических углеводородов (ПАУ), многие из которых являются канцерогенами. Эти вещества определяют в природных и сточных водах, воздухе, почвах, продуктах питания. Тот же метод используют для обнаружения других токсикантов (диоксины, нитрозамины, пестициды), а также многих биологически активных веществ (витамины, гормоны, антибиотики). Люминесцентные детекторы применяют в хроматографическом анализе (см. главу 7), измеряя интенсивность свечения веществ, по очереди выходящих из хроматографической колонки. Люминесцентный анализ применяют в криминалистической экспертизе и для диагностики заболеваний.
Люминесцировать могут далеко не все молекулы, поглощающие свет. Большая часть органических веществ, способных люминесцировать, – это ароматические соединения, имеющие жесткую структуру молекулы.


Фенолфталеин Флуоресцеин
Так, фенолфталеин и флуоресцеин имеют сходное строение молекул (см. схему). Но у фенолфталеина все три бензольных кольца могут свободно колебаться друг относительно друга, и это соединение не люминесцирует. У флуоресцеина же возможность внутримолекулярных колебаний гораздо меньше (кислородный мостик жестко фиксирует два бензольных кольца), и это соединение интенсивно светится в видимой области при облучении его раствора УФ-светом.
Если ароматическое соединение имеет не только жесткую структуру молекулы, но и набор функциональных групп донорного характера (-ОН, -COOH и т. п.), способных к образованию ковалентных связей с ионами металлов, то оно может быть аналитическим реагентом. Для высокочувствительного и селективного определения нелюминесцирующих веществ применяют множество реагентов такого типа. Спектры люминесценции свободного реагента и его комплекса с определяемым ионом различны. Аналитический сигнал, создаваемый комплексом, измеряют на такой длине волны, при которой избыток реагента не люминесцирует. Некоторые неорганические вещества можно определять и по их собственной люминесценции. Так определяют соединения урана, лантаноидов и еще ряда элементов.
Аппаратура для люминесцентного анализа. Для идентификации индивидуальных соединений и для выбора оптимальных условий измерения аналитического сигнала надо изучать спектры возбуждения и спектры испускания люминесценции. С этой целью используют различные спектрофлуориметры. Это более сложные и дорогие приборы, чем ранее рассмотренные спектрофотометры. Принципиальная схема спектрофлуориметра включает: источник возбуждающего света (1), фокусирующую линзу (2), первичный монохроматор (3), входные и выходные щели (4), кюветное отделение (5), вторичный монохроматор (6), приемник люминесцентного изучения (7), регистрирующее устройство (8).

Рис. 6.26. Принципиальная схема спектрофлуориметра
В качестве источников возбуждения чаще всего используют мощные УФ-лампы (ртутные, ксеноновые и др.), а также лазеры. Первичные и вторичные монохроматоры включают дифракционные решетки или призмы, изготовленные из кварца, а также щели регулируемой ширины. Длину волны излучения, выходящего из первичного или вторичного монохроматора (соответственно 1 или 2), можно менять. Первичный монохроматор нужен, чтобы задать оптимальные условия возбуждения определяемого соединения (Х), а также задержать свет лампы с той же длиной волны, что будет испускать Х при люминесценции. Вторичный монохроматор нужен, чтобы создать оптимальные условия регистрации аналитического сигнала Х и не допустить попадания возбуждающего света на фотоприемник. Без этих монохроматоров фоновый фототок оказался бы настолько сильным, что зафиксировать аналитический сигнал Х (люминесцентное излучение) было бы невозможно.
Чтобы получить спектр возбуждения некоторого Х, выставляют на вторичном монохроматоре постоянное значение 2 и меняют величину 1 с помощью первичного монохроматора. Чтобы получить спектр испускания Х, выставляют на первичном монохроматоре постоянное значение 1 и меняют величину 2 с помощью вторичного монохроматора. Значения 1 и 2 , при которых наблюдались максимумы в спектрах возбуждения и испускания Х, в дальнейшем всегда используют при определении этого соединения.
В упрощенных и дешевых приборах для люминесцентного анализа (флуориметрах) вместо монохроматоров используют сменные светофильтры (первичный и вторичный). Приемником люминесценции, как и в спектрофлуориметрах, обычно служат фотоэлемент или фотоумножитель. Фототок усиливают, а затем измеряют с помощью микроамперметра.
Спектры люминесценции. В обычных условиях любые спектры люминесценции индивидуальных веществ являются широкополосными. Спектр возбуждения Х практически совпадает со спектром поглощения того же вещества. Спектры испускания люминесценции похожи на спектры поглощения соответствующих молекул, поскольку также возникают за счет электронных, колебательных и вращательных переходов (рис. 6.27). Однако возбужденные молекулы Х успевают растратить некоторую часть ранее поглощенной энергии (на так называемые безызлучательные переходы, отмеченные на рис. 6.27 волнистыми линиями), прежде чем испустят кванты вторичного излучения. Именно поэтому спектр испускания люминесценции смещен по отношению к спектру поглощения в более длинноволновую область. Эта закономерность известна как правило Стокса–Ломмеля. Чаще всего вещество поглощает возбуждающий свет в УФ-области, а люминесцирует – в видимой.
Как видно из рис. 6.27, механизмы возникновения флуоресценции и особенно фосфоресценции весьма сложны. Необходимые разъяснения следует найти в дополнительной литературе, перечень которой приведен в конце этого учебника.

Рис. 6.27. Схема энергетических переходов, поясняющая возникновение люминесценции
Если вещество способно и флуоресцировать, и фосфоресцировать, то его спектр испускания фосфоресценции сдвинут в длинноволновую сторону еще сильнее, чем спектр флуоресценции (рис. 6.28).

Рис. 6.28. Спектры поглощения (А), флуоресценции (Б) и фосфоресценции (В) одного соединения
Качественный люминесцентный анализ. По появлению люминесценции при УФ-облучении пробы, а также по характерному цвету люминесценции можно визуально опознавать некоторые элементы, например, уран, находящийся в растворе в виде уранил-ионов. Известен целый ряд качественных реакций, приводящих к образованию люминесцирующих соединений. Так, например, можно открывать (а затем и определять) субмикрограммовые количества ионов кадмия по реакции с кальцеином, ионы бериллия и циркония – по реакции с морином, ионы цинка – с салициловой кислотой, ионы алюминия – с 8-оксихинолином. Высокая селективность этих реакций объясняется, во-первых, избирательностью реакции комплексообразования, во-вторых, избирательностью возбуждения люминесценции на используемой длине волны 1, а в-третьих, избирательностью регистрации сигнала – люминесцировать способны далеко не все соединения, поглощающие свет.
По характерному цвету люминесценции выявляют и присутствие некоторых органических соединений. Например, полиароматические углеводороды (ПАУ) дают синее свечение, а смолы и асфальтены – сине-зеленое. Во этих случаях люминесценцию наблюдают визуально, облучая анализируемый образец ультрафиолетовым светом ртутной лампы. Нелюминесцирующие соединения при этом не мешают. Чтобы выявить присутствие одного люминесцирующего соединения в присутствии другого, придется снимать спектр возбуждения или испускания исследуемой пробы и сопоставлять их с атласом соответствующих спектров. Но спектры люминесценции структурно-родст- венных соединений (например, спектры разных ПАУ) весьма близки. Идентифицировать индивидуальные соединения в их неразделенной смеси по обычным (широкополосным) спектрам люминесценции удается крайне редко, спектры разных веществ накладываются друг на друга.
Аналитические возможности люминесцентного анализа существенно расширило одно открытие советских физиков. В 1952 г. было установлено, что спектры люминесценции разбавленных и замороженных растворов ароматических соединений существенно отличаются от обычных спектров люминесценции. Спектр замороженного раствора будет состоять из большого числа очень узких полос, больше похожих на линии. Такие спектры называются квазилинейчатыми. Для их получения надо специально подбирать растворитель и очень сильно охлаждать раствор – до температуры жидкого азота (–196 °С). Данный эффект носит название эффект Шпольского (рис. 6.29).

Рис. 6.29. Спектр люминесценции раствора антрацена в н-гексане при комнатной температуре (справа) и при –196 °С (слева)
Количественный люминесцентный анализ. Интенсивность люминесценции Iлюм пропорциональна числу поглощенных квантов возбуждающего света (Nпогл) и квантовому выходу люминесценции:
Iлюм = K В Nпогл , (6.34)
где К – коэффициент пропорциональности.
Как было установлено С.И. Вавиловым, квантовый выход (B) не зависит от длины волны возбуждающего света (вплоть до некоторого значения крит, специфического для каждого Х). При крит люминесценция Х не наблюдается. Квантовый выход зависит от температуры. С повышением температуры квантовый выход падает, этот эффект называют температурным тушением люминесценции. Квантовый выход вещества Х может также зависеть от рН раствора, если при изменении рН сдвигается равновесие между разными формами Х, а способность к люминесценции у разных форм Х неодинакова (этот эффект проявляется у флуоресцентных индикаторов). Например, флуоресцеин сильнее светится в щелочном растворе. А люминесценция углеводородов не зависит от рН раствора, поскольку они не способны к протолизу.
Используя математическое выражение закона Бугера–Ламберта–Бера в его экспоненциальной форме (см. формулу 6.28), после несложных преобразований можно связать число поглощенных квантов с молярной концентрацией Х в исследуемом растворе:
Nпогл = k I0 (1 – 10–εlc). (6.35)
При условии εlc << 1, Nпогл ≈ 2,3k I0 εlc. Подстановка этого выражения в формулу (6.34) приводит к искомой зависимости интенсивности люминесценции от концентрации:
Iлюм ≈ 2,3k В I0 ε l c. (6.36)
Очевидно, аналитический сигнал Iлюм должен быть прямо пропорционален концентрации Х. Чувствительность определения Х тем выше, чем больше интенсивность возбуждающего света и чем выше квантовый выход люминесценции.
Уравнение (6.36) является основанием для проведения расчетов в количественном люминесцентном анализе. Оно определяет возможность построения градуировочных графиков, использования метода добавок или метода сравнения с эталоном. Однокомпонентные системы изучают с применением флуориметра, многокомпонентные – с применением спектрофлуориметра. Важно, чтобы при измерения сигнала пробы и эталонов все условия сохранялись неизменными (температура, растворитель, рН, толщина слоя раствора в кювете, длины волн или светофильтры при возбуждении и регистрации люминесценции).
Однако условие εlc << 1, обеспечивающее прямолинейность градуировок, выполняется только при очень малых концентрациях Х, обычно до 10–7–10–5 моль/л. В соответствии с формулой (6.35) при больших концентрациях градуировочные графики начинают искривляться, их наклон уменьшается. Одновременно может уменьшаться и квантовый выход люминесценции. Этот эффект называют концентрационным тушением. Причинами являются: увеличение вероятности безызлучательных переходов из-за более частых столкновений молекул Х; образование ассоциатов Х; самопоглощение люминесценции невозбужденными молекулами Х. Концентрационное тушение может приводить не только к искривлению градуировок, но и к снижению сигнала при увеличении концентрации Х (рис. 6.30). Одному и тому же значению сигнала начинают соответствовать две разные концентрации Х. Чтобы устранить неоднозначность результата, можно разбавить раствор и вновь измерить его люминесценцию. Если при этом интенсивность люминесценции уменьшилась, значит, концентрация раствора была равна С1, если увеличилась – С2 . Перечисленные закономерности люминесценции и их физическое объяснение детально описаны в дополнительной литературе.
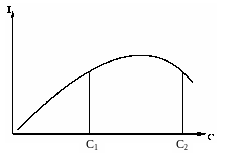
Рис. 6.30. Зависимость интенсивности люминесценции от концентрации Х в условиях концентрационного тушения
Снижение интенсивности люминесценции нередко происходит из-за присутствия посторонних веществ. Механизм влияния примесей может быть химическим или физическим. Химическое взаимодействие между Х и примесью может приводить к образованию соединений, которые при данной длине волны не люминесцируют (или вообще не люминесцируют). Это могут быть окислительно-восстановительные реакции, реакции протолиза, комплексообразования или осаждения. В присутствии некоторых примесей резко снижается квантовый выход, причем тем сильнее, чем выше концентрация этой примеси («гашение люминесценции»). В результате будут получены ошибочные (заниженные) результаты люминесцентного анализа. Вместе с тем, если эффект гашения люминесценции пропорционален концентрации некоторой примеси, это может быть использовано для ее количественного определения.
Физический механизм не обязательно связан со снижением квантового выхода. Нередко влияние примесей объясняется тем, что посторонние вещества поглощают люминесцентное излучение Х.
Метрологические характеристики люминесцентного анализа. Мы уже отмечали основное достоинство люминесцентного анализа – его высокую чувствительность. Так можно определять концентрации порядка 10–8–10–7 г/л, а в отдельных случаях – до 10–12 г/л. Весьма высока и селективность определения люминесцирующих веществ (особенно в условиях эффекта Шпольского). А вот воспроизводимость метода невысока, она значительно хуже, чем воспроизводимость фотометрического анализа (вспомним, сколько трудноконтролируемых факторов влияют на квантовый выход и общую интенсивность люминесценции!). Величина sr здесь обычно не меньше, чем 0,10–0,15. Общая же погрешность результата анализа нередко доходит до 20–30 %. Именно поэтому люминесцентный анализ преимущественно используют там, где нужны высокая чувствительность и селективность, а очень высокая точность не требуется. Например, при оценке загрязнения окружающей среды.