

Зависимость значения коэффициентов c и m от режима течения жидкости
Режим |
Re = Vц d/ |
m |
C |
Ламинарный |
1,6 |
2 |
1,7∙10-4 |
Переходный |
1,6–420 |
1,2 |
2,49∙10-3 |
Турбулентный |
> 420 |
5,36 |
0,5 |
Скорость осаждения частиц на боковую поверхность аппарата пропорциональна квадрату скорости вращения частицы (j = v2/R). В первом приближении скорость осаждения частиц можно принимать равной скорости воды на входе в аппарат.
Когда требуется разделение суспензии и высокая степень обезвоживания осадка, используют центрифугирование. Центробежное фильтрование достигается вращением раствора в перфорированном барабане с мелкой сеткой или тканью. Осадок остается на фильтре на стенках барабана. Уравнение центрифугирования имеет вид
![]() ,
(32)
,
(32)
где V – объем фильтрата за время ; в – плотность жидкости; – динамическая вязкость жидкости; – угловая скорость вращения ротора центрифуги; R – радиус ротора; ro, roc – внутренний радиус жидкости и осадка; L – длина ротора; Кс – коэффициент пропор-циональности слоя.
Контрольные вопросы
Объясните сущность механизма осаждения дисперсных частиц в жидкой среде.
Каковы особенности осаждения частиц в ламинарном, турбулентном и переходном режимах?
Что такое стесненное осаждение?
Особенности механизма отстаивания всплывающих частиц.
Как влияет торможение жидкости на всплытие легких частиц?
Что такое эффект отстаивания?
Объясните механизм фильтрования частиц плоскими перего-родками.
Назовите основные характеристики пористого объемного фильтра.
Каков механизм фильтрования зернистыми перегородками? Общее уравнение процесса фильтрования объемными фильтрами.
Объясните механизм удаления взвешенных частиц под действием центробежных сил.
Уравнение центробежного фильтрования.
Лекция 3. Физико-химические процессы широкого
применения в технологии очистки вод
1. КОАГУЛЯЦИЯ [1, 3, 5, 6]
Коагуляцией называется процесс укрупнения дисперсных частиц в результате их взаимодействия и объединения в агрегаты. Коагуляцию применяют для ускорения процесса осаждения тонкодис-персных примесей и эмульгированных веществ (коллоидно-дисперс-ные частицы размером 3100 мкм).
Коагуляция может происходить самопроизвольно и под влиянием химических и физических процессов. При очистке воды для интенсификации процессов вводят специальные вещества – коагулян-ты. Образующиеся в воде хлопья гидроокисей металлов, имея слабый положительный заряд и взаимодействуя со слабым отрицательным зарядом коллоидных частиц, быстро оседают под действием силы тяжести, увлекая с собой коллоидные и взвешенные частицы примесей.
Коллоидные системы состоят из двух фаз: дисперсионной среды (например, вода) и дисперсной фазы – распределенных в дисперсной среде коллоидных частиц. Коллоидные частицы обладают сложной структурой, образование которой можно представить следую-щим образом: при попадании частицы твердого тела в разбавленный раствор электролита, например в природную воду, частица на своей поверхности адсорбирует из раствора ионы, так называемые потенциал- образующие ионы. Обычно поглощаются ионы, входящие в состав частицы, или другие, близкие по размерам к ионам кристаллической решетки. В результате поглощения ионов поверхность частицы приобретает заряд. В свою очередь находящиеся рядом противоположно заряженные ионы (противоионы) сорбируются у поверхности частицы вследствие электростатического притяжения, образуя коло-идную частицу. В результате ядро коллоидной частицы оказывается окруженным двойным электрическим слоем. Расположенные в непосредственной близости от ядра, противоионы образуют адсорбционный слой противоионов. Несколько дальше от ядра располагаются остальные противоионы, образуя диффузный слой противоионов. Коллоидную частицу вместе с диффузным слоем называют мицеллой (рис. 1, 2).

Рис. 1. Мицеллы хлорида серебра:
1 адсорбционный слой;
2 адсорбционный слой противоионов;
3 диффузионный слой.
аа
б
A Б
А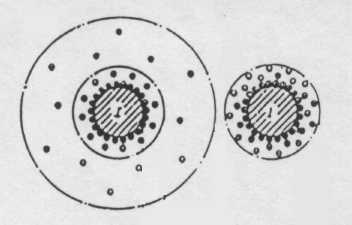



















Рис. 2. Строение мицеллы [6]:
а > 0,03; б = 0;
А адсорбционный слой;
Б диффузионный слой;
I ядро.
Формула мицеллы золя хлорида серебра имеет вид [5]:
![]() ,
(33)
,
(33)
где в фигурных скобках – коллоидная частица; (AgCl)m – ядро коллоидной частицы; m – число молекул хлорида серебра в ядре, n – число адсорбированных ионов Ag+(Cl)-, (n–x) – число противоионов адсорбционного слоя, х – число противоионов диффузного слоя.
В результате теплового движения ионов в растворе часть ионов диффузного слоя отрывается от коллоидной частицы. При этом, между адсорбционным (неподвижным) и диффузным (подвижным) слоями возникает разность потенциалов – так называемый электрокинети-ческий или дзета-потенциал (-потенциал). Этот потенциал обеспе-чивает действие электростатических сил отталкивания частиц. Поэтому коллоидные частицы не слипаются, и коллоидная система остается стабильной. Для того чтобы произошла коагуляция коллоидных частиц, необходимо снизить их дзета-потенциал добавлением в раствор ионов с положительным или отрицательным зарядом.
Коагулирующая способность иона зависит от двух факторов: его валентности и способности адсорбироваться. Очевидно, что двухва-лентный ион нейтрализует в коллоидной частице вдвое больше зарядов противоположного знака, чем одновалентный. Трехвалентные ионы значительно легче адсорбируются на поверхности коллоидной час-тицы, чем одновалентные или двухвалентные. Чем легче адсор-бируется ион, тем меньшее количество его требуется в растворе для достижения адсорбционного равновесия, приводящего к коагуляции.
Наиболее часто в качестве коагулянтов используют сернокислый алюминий (Al2(SO4)3), железный купорос FeSO4, хлорное железо FeCl3 и их смеси.
При поступлении в очищаемую воду, например, сернокислого алюминия происходит его диссоциация[1]
Al2(SO4)3 2Al 3+ + 3SO42- . (34)
Далее идет ионный обмен катионов алюминия на катионы, сорбированные содержащимися в воде частицами. В результате гидролиза оставшихся в избытке катионов алюминия происходит образование выпадающей в осадок гидроокиси алюминия
Al3+ + 3H2O Al(OH)3 + 3H+. (35)
Для нейтрализации отрицательно влияющих на ход реакции катионов водорода, вводят известь или соду. Катионы водорода связы-ваются в воду путем
− добавления извести (гашеная известь – Ca(OH)2)
H+ + OH− H2O; (36)
− добавления соды (кальцинированная сода − Na2CO3):
2H+ + CO32− CO2 + H2O. (37)
В общем виде, процесс гидролиза коагулянтов и образования хлопьев происходит в три стадии [3]:
Me3+ + HOH Me(OH)2+ + H+
Me(OH)2+ + HOH Me(OH)2+ + H+
Me(OH)2+ + HOH Me(OH)3 + H+
Me3+ + HOH Me(OH)3 + 3H+ (38)
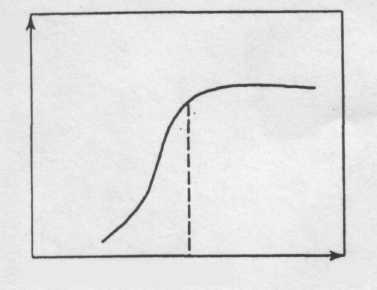
Скорость коагуляции зависит от концентрации электролита (коагулянта). В общем виде эта зависимость показана на рис. 3. При малых концентрациях электролита эффективность соударений частиц (отношение числа столкновений, приведших к слипанию, к общему числу столкновений) близка к нулю, т. е. = 0.
С увеличением концентрации электролита скорость коагуляции возрастает, но не все столкновения приводят к слипанию – такая коагуляция называется медленной. Когда все столкновения приводят к образованию агрегатов ( = 1) наступает быстрая коагуляция [3].
При броуновском движении частиц (неподвижная среда) по теории Смолуховского скорость быстрой коагуляции равна
dnx / d = K (no − nx)2 , (39)
где nx число образующих агрегатов, no начальная концентрация частиц; К константа коагуляции.
0 φ = 1
n
![]()
Рис. 3. Зависимость относительной скорости коагуляции
от концентрации электролита
Количество частиц, образующихся в единице объема воды за время , для быстрой и медленной коагуляции можно определить по формулам:
nб() = no/ (1+/ T1/2), (40)
nм() = no/ [ 1+ (/ T1/2)], (41)
где T1/2 – время уменьшения количества частиц вдвое; – эффектив-ность соударения частиц (определяется из опыта); nо – начальная концентрация частиц.
Для ламинарного и турбулентного движения потока воды число взаимодействий частиц за единицу времени в единице объема жидкости nл и nт вычисляется:
nл = 1/6 n1 n2G (d1+ d2)3, (42)
nт
= 5 n1
n2
R2
![]() ,
(43)
,
(43)
где
R – расстояние, на которое должны
приблизиться частицы, чтобы произошло
слипание (R = 2r); n1
и n2
– число частиц с размером d1
и d2;
G – скоростной градиент (G = dv/dz);
![]() и
и
![]() –
среднеквадратичные скорости двух
коагулирующих частиц.
–
среднеквадратичные скорости двух
коагулирующих частиц.
Из формул видно, что в полидисперсных системах коагуляция проходит быстрее, т. к. более крупные частицы при оседании увлекают за собой более мелкие. Аналогично, удлиненные частицы коагулируют быстрее, чем шарообразные.
2. ФЛОКУЛЯЦИЯ [3]
Для интенсификации процесса коагуляции и повышения скорос-ти осаждения хлопьев в очищаемую воду кроме коагулянтов добавляют высокомолекулярные соединения − флокулянты. Применение флоку-лянтов обеспечивает агрегацию частиц не только за счет непосредст-венного их контакта, но и за счет взаимодействия молекул адсорби-рованного на частицах флокулянта.
Механизм действия флокулянтов основан на следующих явлениях:
адсорбции молекул флокулянта на поверхности коллоидных частиц;
образовании сетчатой структуры из молекул флокулянта;
слипании коллоидных частиц за счет сил Ван-дер-Ваальса (силы притяжения между молекулами).
При действии флокулянта между коллоидными частицами образуются сложные структуры, способные к быстрому осаждению из жидкой фазы. Причиной образования таких структур является адсор-бция макромолекул флокулянта на нескольких частицах с образова-нием между ними перемычек и хлопьев.
Эффективность флокулянта рассчитывают по формуле
ф = (Wсф − W) / Wq, (44)
где Wсф и W – скорость осаждения сфлокулированного и несфлокули-рованного шлама, соответственно, мм/с; q – расход флокулянта на 1 т коагулянта, г.
3. ФЛОТАЦИЯ [3, 6]
Флотация – это процесс удаления из сточной воды нераствори-мых диспергированных гидрофобных (плохо смачиваемых водой) твердых частиц, которые сами плохо отстаиваются. Иногда флотацию используют для удаления растворимых веществ (например, ПАВ). В этом случае процесс называют пенной сепарацией или пенным концентрированием.
Механизм флотации заключается в следующем:
при сближении поднимающегося в воде пузырька воздуха с твердой гидрофобной частицей разделяющая их прослойка воды прорывается и происходит слипание пузырька с частицей;
затем комплекс «частица–пузырек» поднимается на поверх-ность воды, где они собираются пенным слоем с более высокой концентрацией частиц примеси воды, чем их содержание в исходной сточной воде.
Вероятность прилипания пузырька к частице зависит от смачиваемости частицы, которая характеризуется величиной краевого угла (рис. 4).
При закреплении пузырька воздуха на частице образуется граница трех фаз – твердой, жидкой и газообразной. Касательные к поверхности пузырька и частицы образуют угол , называемый краевым углом смачивания.
Энергия образования комплекса «пузырек–частица» А равна:
А = (1– cos ), (45)
где – поверхностное натяжение воды на границе с воздухом; – краевой угол смачивания.
Из формулы видно, чем больше краевой угол , тем больше вероятность прилипания и прочность удержания пузырька на поверхности частицы.

Рис. 4 . Схема элементарного акта флотации:
1 пузырек газа;
2 твердая частица
.
Для хорошо смачиваемых водой частиц 0, а cos 1 и комплекс не образуется; для несмачиваемых частиц прочность прилипания максимальна.
Вероятность образования комплекса «пузырек–частица» зависит от размера и количества пузырьков воздуха, концентрации и размеров частиц и может быть определена по формуле [3]
 ,
(46)
,
(46)
где
n – число пузырьков радиуса R в объеме
V жидкости; r – радиус частицы;
![]() – объемная концентрация газовой фазы.
– объемная концентрация газовой фазы.
Плотность флотационной среды (вода, пузырьки воздуха, твердые частицы) равна
с = ж (1 – Сч – Cг) + ч Сч + г Сг, (47)
где ж, ч, г – плотность жидкости, частиц и газа соответственно; Сч, Cг – объемная концентрация частиц и газа в воде.
Скорость движения частиц vч и пузырьков vп относительно cреды определяется по формулам
vч
![]() [(1
– Cч)
(ч/ж
– 1) + Сг];
(48)
[(1
– Cч)
(ч/ж
– 1) + Сг];
(48)
vп
![]() [1+Cч
(ч/ж
– 1) – Сг],
(49)
[1+Cч
(ч/ж
– 1) – Сг],
(49)
где с – динамическая вязкость флотационной среды; g – ускорение свободного падения (силы тяжести).
Скорость процесса выведения частиц флотацией в общем виде описывается уравнением
dСч / d = – К Сч , (50)
где К – коэффициент скорости флотации, зависящий от гидродинамики и конструктивных параметров установки флотации (определяют опытным путем).
Из опыта установлено, что наилучшее разделение имеет место при соотношении между газообразной и твердой фазами равном Gг/Gч = 0,01–0,1. Это соотношение находят по формуле
Gг/Gч =1,3b(fР−1)Q1/Сч Q, (51)
где Gг и Gч – масса воздуха и твердого вещества, соответственно, г; b – растворимость воздуха в воде при атмосферном давлении и данной температуре, см3/л; f – степень насыщения (f = 0,5–0,8); P – абсолютное давление, при котором вода насыщается воздухом; Q1 – количество воды, насыщенной воздухом, м3/ч; Q – расход сточной воды, м3/ч.
Контрольные вопросы
Объясните механизм процесса коагулирования. Приведите примеры реакций применения коагулянтов.
Напишите общее уравнение скорости коагуляции. В чем разница между быстрой и медленной коагуляцией.
Напишите уравнения для скорости коагуляции при ламинарном и турбулентном движении потока жидкости.
Что такое флокуляция? Каков механизм этого процесса?
Объясните механизм элементарного акта флотации.
Напишите уравнения, характеризующие основные парамет-ры флотации: вероятность образования комплекса «пузырек–частица», скорость движения пузырьков и частиц в среде.
Общее уравнение процесса выведения частиц флотацией.
Лекция 4. Физико-химические процессы, применяемые
при очистке сточных вод от растворенных
органических веществ
1. ПЕННАЯ СЕПАРАЦИЯ [3]
Пенное концентрирование (сепарация) основано на селективной адсорбции веществ поверхностью газовых пузырьков, которые через раствор поднимаются на поверхность раздела фаз. Парциальную сепа-рацию отдельных компонентов раствора обеспечивает образующаяся в растворе пена. Этот процесс используют для удаления из сточных вод ПАВ (барботаж воды с ПАВ).
Адсорбция органических веществ на поверхности раздела фаз «газ–жидкость» связана с изменением поверхностного натяжения и с избыточной поверхностной концентрацией удаляемого вещества уравнением
d = Гi d i , (52)
где
d
– изменение поверхностного натяжения;
Гi
– избыточная концентрация вещества на
поверхности; i
– химический потенциал i-й составляющей
(![]() ,
где R – газовая постоянная
,
где R – газовая постоянная
![]() ,
лат/градмоль;
ai –
термодинамическая активность, которая
при больших разбавлениях раствора равна
концентрации растворенного вещества
сi).
,
лат/градмоль;
ai –
термодинамическая активность, которая
при больших разбавлениях раствора равна
концентрации растворенного вещества
сi).
Отсюда коэффициент распределения Кi (отношение концентраций в двух фазах) равен
![]() .
(53)
.
(53)
Для разбавленных растворов d/ dсi практически не зависит от концентрации и Кi становится постоянным. Повышение расхода возду-ха приводит к увеличению частоты образования пузырьков и росту объема пены. Соответственно растет количество ПАВ, адсорбирую-щихся на поверхности раздела фаз. Кинетика извлечения ПАВ определяется уравнением
![]() ,
(54)
,
(54)
где d/ dск – изменение поверхностного натяжения в зависимости от остаточной концентрации растворенных в объеме воды ПАВ.
При линейном изменении величины поверхностного натяжения воды от концентрации ПАВ второй член стремится к нулю, и уравнение кинетики процесса принимает вид:
ln ск / со = –К, (55)
где ск – остаточная концентрация ПАВ в объеме воды; со –концентрация ПАВ в момент времени о (при о = 0; ск = со); – время; К – константа (из опыта).
Степень извлечения ПАВ пеной равна
п = 100 (сн – ск) /сн = сп /сн , (56)
где сн – начальная концентрация ПАВ в воде, сп – концентрация ПАВ в пене.
Коэффициент распределения ПАВ между пеной (пеноконденса-том) и сточной водой, характеризующий эффективность извлечения ПАВ, равен
п = сп /ск. (57)
В практике очистки сточных вод от ПАВ стремятся, чтобы пеноконденсат имел наименьший объем с максимальной концент-рацией ПАВ. Коэффициент распределения п всегда больше единицы.
2. АДСОРБЦИЯ [3,7]
Адсорбция применяется для глубокой очистки сточных вод от растворенных органических веществ (гербицидов, пестицидов, ПАВ, красителей и других органических веществ). Метод адсорбции обладает высокой (до 90 %) эффективностью очистки. Возможна очистка одновременно от нескольких веществ, а также рекуперация этих веществ.
Физическая сущность адсорбции заключается в способности твердого сорбента удерживать загрязняющее вещество под действием силового поля поверхности. В зависимости от характера взаимо-действия между молекулами адсорбента и поглощаемого вещества (адсорбата) различают физическую адсорбцию и хемосорбцию. Физии-ческая адсорбция менее прочна, не сопровождается существенным изменением (в отличие от хемосорбции) молекул адсорбента. Она обус-ловлена силами межмолекулярного взаимодействия, то есть такими, которые связывают молекулы в жидкостях или газах (ионная связь между противоположно заряженными ионами, ковалентная связь меж-ду электронейтральными атомами). Физически адсорбированные моле-кулы достаточно свободно перемещаются по поверхности сорбента. Они могут собираться группами и через некоторое время могут покидать поверхность адсорбента это десорбция. Время, в течение которого молекула находится на поверхности адсорбента, называют временем адсорбции. Скоростью адсорбции (десорбции) называют количество молекул адсорбирующихся (десорбирующихся) за единицу времени на единице поверхности или массы адсорбента. Если скорости адсорбции и десорбции равны между собой, то говорят, что наступило адсорбционное равновесие. Оно может оставаться постоянным, если остаются постоянными внешние условия (давление, температура, концентрация). С увеличением концентрации или давления пропорционально возрастает скорость адсорбции и увеличивается равновесное количество адсорбированных молекул. С ростом температуры уменьшается время адсорбции и равновесное количество адсорбированных молекул. Кривые зависимости равновесной адсорбции от концентрации или давления адсорбата при постоянной температуре называются изотермами адсорбции.
Для описания равновесного состояния процесса сорбирования в воде с одним растворенным веществом используют уравнения изотерм Ленгмюра qe = kc/(1+bc) или Фрейндлиха qe = Kc1/n, где qe – равновесная величина адсорбции, связанная с концентрацией с; b – коэффициент, зависящий от температуры (определяют эксперименталь-но); k, K и n – соответствующие постоянные для данной температуры коэффициенты.
Фазовая диаграмма в виде изотермы сорбции является одним из основных критериев оценки адсорбционных свойств сорбента.
Вещества, хорошо адсорбируемые из водных растворов сорбентом, имеют обычно выпуклую изотерму адсорбции, а плохо сорбирующиеся вогнутую (рис. 5) [7].
Благоприятный
процесс
Неблагоприятный
процесс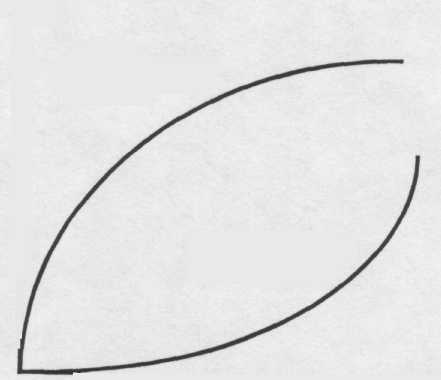



Концентрация в
растворе при равновесии, г/л
Рис. 5. Вид фазовой диаграммы сорбционного равновесия

Изотерму адсорбции вещества, находящегося в сточной воде, получают опытным путем. Она позволяет определить:
условия максимального сорбирования загрязняющего вещества даже при низкой его концентрации;
условия, при которых на удаление небольшого количества заг-рязняющей примеси потребуется большое количество сорбента и, соответственно, значительные затраты на его регенерацию;
границы процесса и предельную сорбционную способность;
насколько реальным является получение продукта требуемой концентрации на выходе.
Изотерму адсорбции можно получить и расчетным путем. В этом случае для различных значений заполнения адсорбционного объема ( = aVi*/Vа) требуется рассчитывать величину равновесной концент-рации и удельной адсорбции. Равновесную концентрацию вычисляют по формуле [3]
 ,
(58)
,
(58)
где
– степень заполнения адсорбционного
объема; а – удельная сорбция;
![]() – молярные объемы извлекаемого вещества
и воды; Кi –
константа адсорбционного равновесия;
fi
– парциальный коэффициент активности
извлекаемого компонента (при малой
величине заполнения адсорбционного
объема fi
= 1).
– молярные объемы извлекаемого вещества
и воды; Кi –
константа адсорбционного равновесия;
fi
– парциальный коэффициент активности
извлекаемого компонента (при малой
величине заполнения адсорбционного
объема fi
= 1).
Величины молярных объемов компонентов раствора (сточной воды) вычисляют по формуле
![]() ,
(59)
,
(59)
где Мi – молекулярная масса; i – плотность компонента.
Константу адсорбционного равновесия определяют из уравнения
![]() ,
(60)
,
(60)
где
![]() –
молярное уменьшение свободной энергии
адсорбции вещества (находят
экспериментально).
–
молярное уменьшение свободной энергии
адсорбции вещества (находят
экспериментально).
Удельную адсорбцию (моль/г) вычисляют по формуле
![]() ,
(61)
,
(61)
где Va – предельный адсорбционный объем пор адсорбента, см3/г.
Если в очищаемой воде имеются несколько компонентов, то для определения возможности их совместного удаления (адсорбции) для каждого вещества находят значение стандартной дифференциальной свободной энергии и определяют разность между максимальным и минимальным значением. Если (ΔF ºmax ΔF ºmin) ≤ 10,5 кДж/моль, то совместная адсорбция возможна. В противном случае очистку нужно проводить раздельно.
В общем случае процесс адсорбции состоит из трех стадий:
перенос компонента из сточной воды к поверхности зерен сорбента (внешняя диффузия);
непосредственно адсорбционный процесс;
перенос вещества внутри зерен адсорбента (внутренняя диф-фузия).
Лимитирующей стадией может быть внешняя или внутренняя диффузия, иногда обе вместе.
Во внешнедиффузионной области скорость массопереноса определяется интенсивностью турбулентности потока и зависит от скорости жидкости.
Во внутридиффузионной области интенсивность массопереноса зависит от вида и размеров: пор сорбента, зерен сорбента, молекул сорбируемого вещества.
Для оптимальной скорости очистки необходимо процесс прово-дить при таких гидравлических режимах, чтобы он лимитировался внутренней диффузией, сопротивление которой можно регулировать изменением структуры сорбента. В расчетах рекомендуется принимать скорость жидкости = 1,8 м/ч, диаметр зерна dз = 2,5 мм. При меньших значениях этих величин процесс лимитируется внешней диффузией, при больших – внутренней.
3. ЭКСТРАКЦИЯ [6]
При смешивании двух взаимно нерастворимых жидкостей любое вещество, находящееся в этой смеси, распределяется между ними в соответствии со своей растворимостью согласно закону распределения. Так, если к сточной воде прибавить нерастворимую в ней органичес-кую жидкость, то находящиеся в воде примеси будут растворяться в прибавленной жидкости, а концентрация их в воде будет умень-шаться. Если же после этого прибавленную жидкость выделить из сточной воды, то последняя окажется частично очищенной от растворенных примесей. Такой физико-химический способ удаления веществ из воды называют жидкостной экстракцией, удаляемые при этом вещества – экстрагируемыми веществами, а добавляемую не смешивающуюся со сточной водой жидкость – экстрагентом. Отношение взаимно уравновешивающихся концентраций в двух несмешивающихся растворителях при достижении равновесия является постоянным и называется коэффициентом распределения:
kp = Cэ/ Сст. const, (62)
где Cэ и Сст. – концентрация экстрагируемого вещества в экстрагенте и в сточной воде при установившемся равновесии.
Коэффициент распределения kp зависит от температуры экстракции и наличия примесей в воде. Сконцентрированное в экстрагенте вещество отделяется от растворителя и может быть использовано.
Для успешного протекания процесса экстракции необходимо, чтобы экстрагент:
имел хорошую экстракционную способность по отношению к экстрагируемому веществу, т.е. имел большой коэффициент распре-деления kp;
имел хорошую селективность;
имел малую растворимость в воде;
имел плотность, отличающуюся от плотности воды;
был неогнеопасен, нетоксичен, химически неактивен;
имел низкую стоимость и др.
Метод экстракционной очистки выгоден при больших (более 2 г/л) концентрациях органических веществ в сточной воде и высокой их стоимости. В качестве экстрагентов обычно используют бензол, бутилацетат, изобутилацетат, гексан и др.
При однократной экстракции не удается полностью удалить из сточной воды экстрагируемое вещество. Поэтому применяют много-кратную экстракцию той же воды. Разработано несколько методов экстрагирования органических веществ. По схемам контакта экстраген-та и сточной воды их можно разделить на перекрестноточные, ступен-чато-противоточные (рис. 6, а, б) и непрерывно-противоточные (рис. 7).
В ступенчато-противоточной схеме экстракции конечная концентрация экстрагируемого вещества в воде может быть определена по формуле:
Ск = Сн / (1+ b kр)n , (63)
где Ск и Сн – конечная и начальная концентрация экстрагируемого вещества в воде; n – число экстракционных узлов; b – удельный расход экстрагента (м3/ м3):
b = V/ (nQ), (64)
где V – общий объем экстрагента, затрачиваемого на экстракцию; Q – количество сточной воды, подаваемой на экстракцию.
В
схеме непрерывно-противоточной экстракции
вода и экстрагент движутся навстречу
друг другу в одном аппарате. Если
плотность сточной воды больше плотности
экстрагента
![]() ст
э,
то вода вводится в экстракционную
колонну сверху, а экстрагент – снизу
(рис. 7, a). При
ст
э
экстрагент вводится в верхнюю часть
колонны, а обрабатываемая вода
ст
э,
то вода вводится в экстракционную
колонну сверху, а экстрагент – снизу
(рис. 7, a). При
ст
э
экстрагент вводится в верхнюю часть
колонны, а обрабатываемая вода![]()
в нижнюю (рис. 7, б). В обработанной воде
при этом содержание экстрагируемого
вещества определяется по формуле
в нижнюю (рис. 7, б). В обработанной воде
при этом содержание экстрагируемого
вещества определяется по формуле
Ск = Сн (1 bkp). (65)
Требуемый удельный расход экстрагента при заданных начальной и конечной концентрациях экстрагируемого вещества в сточной воде определяется:
![]() .
(66)
.
(66)
Рис. 6. Схема
процесса многоступенчатой экстракции:
а)
перекрестноточная;
б)
ступенчато-противоточная;
1 – трубопровод
для подачи сточной воды; 2, 3 и 4 –
экстракционные установки I,
II, III
ступеней; 5 – трубо-провод для подачи
чистого экстрагента; 6 – трубопровод
для выпуска обработанной сточной воды;
7 – трубопровод для выпуска отработанного
экстрагента; 8 – отстойник (или сепаратор)
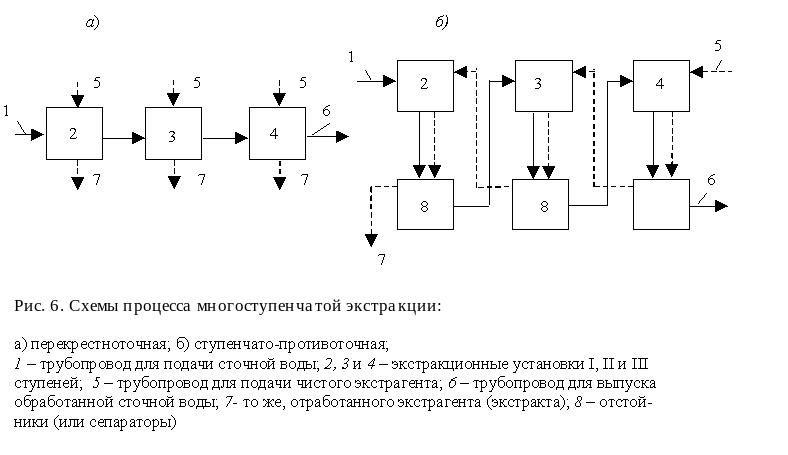
Рис.
7. Схема процесса непре-рывно-противоточной
экстрак-ции:
а)
с подачей сточной воды сверху, экстрагента
– снизу; б) с подачей сточной воды снизу,
экстрагента – сверху;
1
– трубопровод для подачи экст-рагента;
2 – трубопровод для подачи сточной
воды; 3 – трубо-провод для выпуска
отработан-ного экстрагента; 4
трубопровод для выпуска отработанной
сточ-ной воды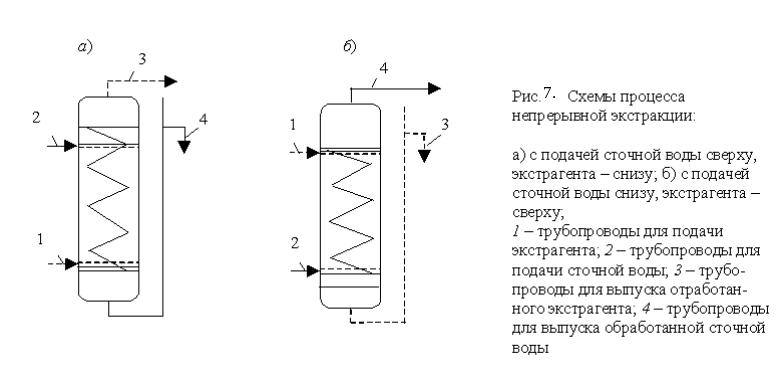
Контрольные вопросы
Что такое пенная сепарация? Дайте определение коэффи-циента распределения.
Напишите общее уравнение кинетики процесса пенной сепарации.
Объясните физическую сущность процесса адсорбции при очистке вод.
Что такое фазовая диаграмма сорбционного равновесия? Ее интерпретация.
Напишите формулы для оценки равновесной концентрации и удельной адсорбции.
Назовите лимитирующие стадии процесса адсорбции и условия для оптимальной скорости очистки.
Объясните сущность процесса экстракции, основные требования при выборе экстрагента.
Назовите основные схемы процессов экстракции и принципы их работы; напишите формулы для оценки концентрации экстраги-руемого вещества в очищенной воде.
Лекция 5. Физико-химические процессы, используемые при очистке вод от металлов, их солей и летучих примесей
1. ИОННЫЙ ОБМЕН [3, 8]
Ионный обмен или ионная сорбция − это процесс обмена между ионами, находящимися в растворе, и ионами, присутствующими на поверхности твердой фазы, называемой ионитом. По знаку заряда обменивающихся ионов, иониты делят на катиониты (которые способны из раствора поглощать положительные ионы) и аниониты (которые поглощают отрицательные ионы). Первые обладают кислотными свойствами, вторые – основными.
Различают следующие виды ионитов:
сильно кислотные катиониты – содержат сульфогруппы SO3H (например, катионит смола КУ-2, основа матрицы – полистирол, бывают ионные формы Н+ и Na+) или фосфорнокислые группы [РО(ОН)2];
слабокислые катиониты – содержат карбоксильные (СООН) группы (например, катионит – смола КБ-1, основа матрицы – метакриловая кислота, ионная форма Na+) или фенольные (С6Н5ОН);
сильноосновные аниониты, содержащие аммониевые основа-ния R3NOH (основа матрицы, например, анионита АВ-17 – полисти-рол);
слабоосновные аниониты, содержащие аминогруппы различной степени замещения (первичные NH2; вторичные = NН и третичные N), в качестве основы матрицы используется полистирол, например, в анионите АН-22 (анионит низкоосновной, функциональная группа – NH2 или RNH).
Важнейшим свойством ионитов является их поглотительная способность, так называемая обменная емкость. Полная емкость ионита – количество находящихся в сточной воде грамм-эквивалентов ионов, которое может поглотить 1 м3 ионита до полного насыщения; рабочая емкость ионита – количество находящихся в воде грамм-эквивалентов ионов, которое может поглотить 1 м3 ионита до начала проскока.
Если катиониты находятся в Н, Na или NH4−форме, обмен катионов будет проходить по реакции:
Ме+ + Н[R] Ме+ [R] + H+ (Н-катионирование), (67)
Ме+ + Na[R] Ме+ [R] + Na+ (Na-катионирование), (68)
Ме+ + NН4[R] Ме+ [R] + NН4+ (NН4-катионирование), (69)
где Ме+ – катион, находящийся в сточной воде; [R] – сложный комплекс катионита (органический скелет), практически нераство-римый в воде, называют матрица. При сокращенном написании ионита матрицу обозначают в виде [R], а активную группу пишут полностью. Например, сульфокатиониты записывают как RSO3H, где R – матрица, Н – обменивающиеся ионы (называют противоионы); SO3 – анкерный ион (связывает противоионы с противоположно заряжен-ными ионами матрицы).
Реакция ионного обмена является обратимой и идет до установ-ления ионообменного равновесия. Обратимость реакции позволяет осу-ществлять регенерацию ионита. Регенерация, например, Н-катионита, использованного ранее для очистки (смягчения) питьевой воды, дости-гается пропусканием через него раствора серной или соляной кислоты:
![]() →
→![]() ,
(70)
,
(70)
т. е. задержанные катионитом ионы Ca или Mg при регенерации кислотой замещаются водородными ионами кислоты.
Процесс переноса вещества при ионном обмене можно представить в виде нескольких стадий (рис. 8):
перенос иона Ме+ из потока жидкости к поверхности жидкой пленки, окружающей зерно ионита;
диффузия ионов через пленку (пленочная диффузия);
переход иона через границу раздела фаз в зерно смолы;
диффузия ионов Ме+ внутри зерна смолы (гелевая диффузия);
химическая реакция двойного обмена ионов Ме+ и Н+ (или другие);
диффузия ионов Н+ (или друге) внутри зерна ионита к границе раздела фаз;
переход ионов Н+ (или другие) через границу раздела фаз на внутренней поверхности пленки жидкости;
диффузия ионов Н+ (или другие) через пленку жидкости;
диффузия ионов Н+ (или другие) в жидкость.
Рис. 8. Схема
переноса вещества при ионном обмене
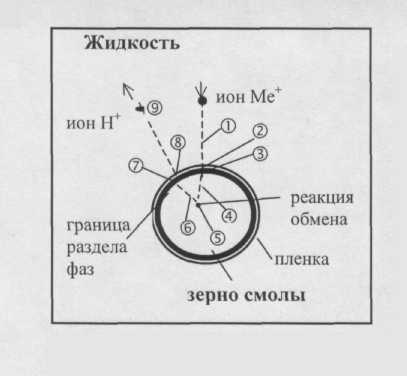
Скорость ионного обмена определяется самыми медленными стадиями – диффузией в зерне ионита (гелевая диффузия) или диффузией в неподвижном слое (пленке) жидкости, окружающей зерно ионита (пленочная диффузия). В некоторых случаях, когда скорости гелевой и пленочной диффузии сравнимы по величине, обе стадии могут контролировать ионный обмен. Согласно пленочной модели допускается существование полностью неподвижной пленки жидкости на поверхности зерна ионита (т. н. пленка Нернста). Толщина пленки Нернста может быть определена по следующей формуле [8]:
= 2ro / Sh, (71)
где – толщина пленки, см; ro – радиус зерна, см; Sh – число Шервуда в безразмерных единицах (определяют экспериментально).
Количественным критерием, позволяющим определить стадию, которая будет контролировать скорость процесса в целом, являются следующие уравнения кинетики ионного обмена:
при гелевой диффузии:
![]() ;
(72)
;
(72)
при пленочной диффузии:
![]() ,
(73)
,
(73)
где
![]() и С – концентрация противоионов в ионите
и жидкости (мг-экв/мл);
и С – концентрация противоионов в ионите
и жидкости (мг-экв/мл);
![]() и D – коэффициенты диффузии (см2/с)
в ионите и жидкости;
и D – коэффициенты диффузии (см2/с)
в ионите и жидкости;
![]() – коэффициент селективности
(избирательности), безразмерная величина.
– коэффициент селективности
(избирательности), безразмерная величина.
Другим подходящим методом для математического описания кинетики ионного обмена является метод линейных движущих сил. Концепция этого метода заключается в том, что скорость процесса ионного обмена предполагается быть пропорциональной степени отклонения системы от равновесного состояния. При этом уравнения для пленочной и гелевой диффузии имеют вид [8]:
![]() и
и
![]() ,
(74)
,
(74)
где
СА
– концентрация ионов А в растворе при
достижении равновесия с ионитом, в
котором средняя концентрация ионов А
равна
![]() (количество ионов в зерне деленное на
объем зерна);
(количество ионов в зерне деленное на
объем зерна);
![]() – равновесная концентрация ионов А в
ионите при концентрации этих ионов в
растворе СА;
kf и
kp
– коэффициенты движущей силы в пленке
и зерне (с–1).
– равновесная концентрация ионов А в
ионите при концентрации этих ионов в
растворе СА;
kf и
kp
– коэффициенты движущей силы в пленке
и зерне (с–1).
Коэффициенты kf и kp можно выразить через соответствующие характеристики зерна, толщину пленки и коэффициенты диффузии в виде
kf = 3D/ro ; kp 0,07 ro2, (75)
где – эффективный коэффициент диффузии в ионите (величина постоянная), см3/с; ro – радиус зерна; − толщина пленки.
Основным недостатком этого метода является допущение, что эффективный коэффициент диффузии есть величина постоянная; при ионном обмене это не всегда верно.
Для определения ионного равновесия используют изотермы ионного обмена, которые представляют собой зависимость противо-ионного состава ионита от противоионного состава раствора при t и Р = const. В безразмерных координатах изотермы для различных фаз системы имеют вид [3]:
![]() и
и
![]() , (76)
, (76)
где
![]() и аi
– эквивалентные доли i-го иона в фазе
ионита и в растворе, соответственно;
и аi
– эквивалентные доли i-го иона в фазе
ионита и в растворе, соответственно;
![]() и Сi
– концентрации i-го иона в ионите и
растворе при равновесии системы, моль
ионов/г ионита; zi
– заряд i-го иона.
и Сi
– концентрации i-го иона в ионите и
растворе при равновесии системы, моль
ионов/г ионита; zi
– заряд i-го иона.
Отношение / аi = Кpi называют коэффициентом распределения i-го иона при сорбции. Этот коэффициент является мерой обогащения ионита. При Кpi < 1 ионит обеднен, а при Кpi > 1 обогащен компонентом i по сравнению с равновесным раствором. В сложных растворах сорбция может проходить по разному для различных ионов, т. е. может наблюдаться селективность ионита по отношению к отдельным ионам. Отношение коэффициентов распределения конкурирующих ионов называют коэффициентом селективности (избирательности).
Например, при наличии в растворе двух ионов А и В коэффициент селективности можно записать следующим образом:
КАВ
= КрА/КрВ
=
![]() аВ/
аА
аВ/
аА![]() =
=
![]() СВ/
СА
СВ/
СА![]() .
(77)
.
(77)
При КАВ > 1 ионит селективен к иону А; при КАВ < 1 избира-тельно сорбируется ион В; при КАВ = 1 ионит не проявляет селективности к обоим ионам.
От величины коэффициента селективности зависит форма изо-термы ионного обмена. При КАВ > 1 изотерма выпуклая, при КАВ < 1 – вогнутая, а при КАВ = 1 – линейная.
2. ОБРАТНЫЙ ОСМОС И УЛЬТРАФИЛЬТРАЦИЯ [3]
Обратным осмосом и ультрафильтрацией называют процессы фильтрования растворов через полупроницаемые мембраны под давлением, превышающим осмотическое (рис. 9).
Мембраны пропускают молекулы растворителя (воду) и задержи-вают растворенные вещества. При обратном осмосе отделяются молекулы, размеры которых меньше размеров молекул растворителя. При ультрафильтрации размер отдельных частиц d на порядок больше (табл. 4).
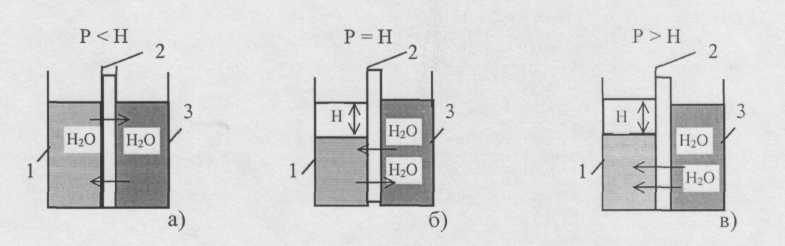
Рис. 9. Схемы осмоса:
а) прямой осмос; б) осмотическое равновесие;
в) обратный осмос;
Н – осмотическое давление; Р – рабочее давление;
1 чистая вода; 2 мембрана; 3 раствор
Таблица 4