

Синдром Ангельмана
СА впервые описан в 1965 г. под названием синдрома «счастливой куклы». Основные признаки: микробрахицефалия с уплощенным затылком, большая нижняя челюсть, открытый рот, выступающий язык, макростомия, редкие зубы, гипопигментация.
У больных детей наблюдается задержка психомоторного развития, невозможность обучения, явления атаксии, гиперкинезия, мышечная гипотония, судорожная готовность и гиперрефлексия. Постоянны приступы неконтролируемого смеха и хлопания в ладоши.
В основе синдрома лежат те же причины, что и при СПВ, однако затронута не отцовская, а материнская хромосома.
Первая причина - варианты делеции критического района материнской хромосомы 15 (15q11.2-q13), возникающие в гаметогенезе в результате внутри- и межхромосомных перестроек.
Вторая причина - ОРД отцовской хромосомы 15, возникающая при нерасхождении хромосом в оогенезе и зависящая от возраста матери на момент рождения ребенка.
Третья причина - ошибки в ЦИ. Генами-кандидатами СА являются следующие.
• Ген UBE3A - основной ген-кандидат, мутации в котором обусловливают развитие 20% случаев СА. Этот ген кодирует фермент У6-АР убиквитин-протеинлигазу, экспрессия которой обнаружена во всех тканях. Кодируемый белок служит коактиватором для рецепторов стероидных гормонов. В ряде структур головного мозга ген UBE3A активен только в материнской хромосоме, что может объяснить развитие атаксии и тремора дефицитом кодируемого геном фермента в клетках Пуркинье, а развитие эпилептических припадков и невозможность обучения ребенка - отсутствием экспрессии этого гена в нейронах гиппокампа. Ген UBE3A транскрибируется с нескольких промоторов и имеет 7 нетранслируемыхэкзонов со стороны 5'-конца. Среди мутаций данного гена преобладают мутации со сдвигом рамки считывания и нонсенсмутации. Описаны также инверсии с точками разрывов внутри
|
гена UBE3A.
• Ген АТР1 С - кодирует аминофосфолипидтранспортирующуюАТРазу. Экспрессируется преимущественно с материнского аллеля в фибробластах кожи и в разных структурах мозга. Передает сигналы в ЦНС и поддерживает контакты между клеточными мембранами. Отсутствие продуктов экспрессии этого гена в мозге больных СА ведет к тяжелому детскому аутизму.
• Ген GABRB3 является одним из генов кластера ГАМКА- рецепторных генов, расположенных между геном UBE3A и локусом Р. Эти рецепторные гены попадают в область наиболее частых делеций критического района. Ген GABRB3 кодирует бета-3-субъединицу ГАМКА-рецептора. Поэтому симптоматика СА (моторные нарушения, нарушения памяти, неспособность к обучению, судорожные припадки) может быть обусловлена гаплонедостаточностью данного гена.
Синдром Беквитта-Видемана
Синдром Беквитта-Видемана (СБВ) относится к распространенным наследственным заболеваниям с частотой в популяции 1:10- 12 тыс. Тип наследования болезни аутосомно-доминантный с неполной пенетрантностью и варьирующей экспрессивностью.
Примерно 15% всех случаев СБВ расцениваются как семейные формы.
Основные клинические признаки СБВ: гигантизм (масса тела при рождении свыше 3900 г), черепно-лицевой дисморфизм (долихоцефалия, гипоплазия верхней челюсти и средней трети лица, прогнатизм, макроглоссия, вертикальные насечки на мочках и небольшие полулунные ямочки на задней поверхности завитков ушных раковин), гемангиомы на лбу, пигментные пятна на коже затылка и лица, пупочная грыжа. У всех больных наблюдается вицеромегалия внутренних органов или увеличение размеров гонад, надпочечников, печени, поджелудочной железы, предстательной железы, почек и селезенки. Предполагается, что вицеромегалиясвязана с сохранением паракринной и эндокринной функций частью эмбриональных клеток на пренатальном и постнатальном этапах онтогенеза. У 15% больных с СБВ выявляется УО. У многих больных наблюдается повышенная предрасположенность к развитию онкогенных заболеваний, например частота нефробластомы достигает 59%, карциномы надпочечников - 15%, гепатобластомы - 2%.
|
Важный диагностический признак - гемигипертрофия, наблюдаемая в 12,5% случаев СБВ, а у больных с опухолями этот признак проявляется в каждом втором случае.
Критической областью для СБВ является терминальная часть короткого плеча хромосомы 11 (11р15.5). В этой области расположен кластер импринтированных генов, перемешанных с неимпринтированными генами (рис. 2).
Критическая область включает в левой части (по отношению к центромере) 6 генов и ограничена геном NAP2; в правой части - 7 генов и ограничена геном L 23.
Кластер генов критической области хромосомы 11(11р15.5) начинается с первого гена - NAP2, биаллельноэкспрессирующегося в эмбриональных и зрелых тканях.
Второй ген - IPL/TSSC3 - преимущественно экспрессируется с материнскогоаллеля и ограниченно - с отцовского аллеля; наиболее активен в плаценте, легких, миокарде, печени, поджелудочной железе и почках. В клетках мозга взрослых людей и лимфоцитах крови он экспрессируетсябиаллельно. При развитии опухолей головного мозга ген IPL/TSSC3, по-видимому, выполняет роль гена-супрессора материнского аллеля.
Третий ген - IMPTI/TSSC5 - кодирует белок мембранного переносчика, контролирующего полиспецифический транспорт и множественную лекарственную устойчивость.
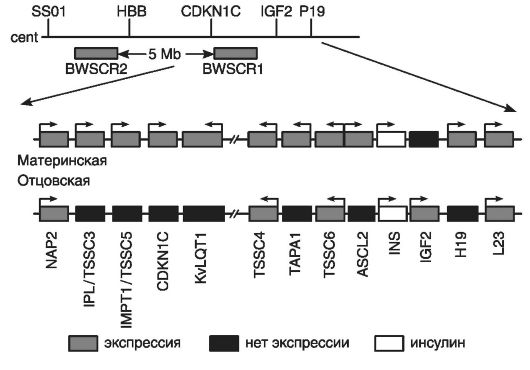
Рис. 2. [3] Схема молекулярной организации критической области 11р15.5 (по Немцовой М.В. и Залетаеву Д.В., 2004)
Кроме того, точковые мутации этого гена и потеря его материнской копии были идентифицированы при раке грудной железы, нефробластоме и рабдомиосаркоме.
С антисмысловой цепи ДНК транскрибируется другой ген этого локуса - ген IMPTI-AS, имеющий множество сайтов инициации транскрипции, но экспрессирующий только два транскрипта. Считается, что этот ген не транслируется и поэтому не относится к импринтированным генам.
|
Четвертый ген - CDKN1C - также известен как ген р57К1Р2. Он кодирует ингибитор циклинзависимойкиназы, относящийся к CIP-зависимым регуляторам клеточного цикла. Его гиперэкспрессия останавливает клеточный цикл в предсинтетической стадии.
В гене обнаружены точковые мутации, в связи с чем он считается реальным геном-кандидатом синдрома СБВ. Мутации гена р57 KIP
встречаются в 40% семейных и 5% спорадических случаев СВБ. В самом гене суммарная частота мутаций составляет 10-15%.
В клетках таких опухолей, как карцинома надпочечников, нефробластома и рабдомиосаркома, выявляется гиперэкспрессия гена р57 KIP с материнского аллеля.
Роль пятого гена - KvLQT1 - в этиологии СБВ точно не определена, однако считается, что он участвует в процессе импринтинга при этом заболевании. Данный ген кодирует белок, формирующий заслонку калиевых каналов. Он экспрессируетсябиаллельно только в сердечной мышце; его точковые мутации сопровождаются доминантно наследуемым дефектом проводимости сердечной мышцы или LQT-синдромом (удлиненный Q-Т-интервал и желудочковая аритмия), а также аутосомно-рецессивным синдромом Джервила-Ланге-Нильсона, сочетающимся с аналогичными нарушениями проводимости миокарда и глухотой. Ген этого синдрома (KvLQT1) экспрессируется с материнского аллеля и обнаруживается в большинстве тканей организма, но он почти не экспрессируется в мозге и скелетных мышцах. В интроне гена KvLQT1 обнаружен ген LIT1, транскрибирующийся с антисмысловой цепи, но только с отцовской хромосомы.
При СБВ установлена биаллельная экспрессия этого гена, тогда как при нефробластоме наблюдается лишь моноаллельная его экспрессия.
Шестой ген TSSC4 и восьмой ген TSSC6 не относятся к импринтированным генам СБВ, но они кодируют белки с опухоль-подавляющей функцией, например белки с предполагаемой опухоль-подавляющей функцией в случае рабдомиосаркомы. Между этими генами расположен седьмой ген - ТАРА1 (CD81), кодирующий интегральный мембранный белок, обнаруженный в разных типах клеток. Этот ген участвует в развитии на ранних этапах Т-лимфоцитов. Он экспрессируется только с материнской хромосомы.
|
Девятый ген - ASL2, также экспрессирующийся с материнскогоаллеля. Его белковый продукт является фактором транскрипции, содержащим мотив: спираль-петля-спираль. Этот белок участвует в развитии трофоэктодермы.
Десятый ген - INS (ген инсулина) имеет биаллельную экспрессию, а находящийся рядом с ним инсулиноподобный фактор роста IGF2 кодирует фетальный фактор роста.
Одиннадцатый ген или IGF2-фактор экспрессируется только с отцовского аллеля, может быть митогеном для всех типов клеток, но у человека он специфически модулирует рост и дифференцировку мышечных клеток. Нарушение экспрессии IGF2 обусловливает органомегалию при СВБ. Биаллельную экспрессию IGF2 обнаруживают в нефробластомах, карциномах коры надпочечников, рабдомиосаркомах, других эмбриональных опухолях, встречающихся при СБВ.
Двенадцатый ген Н19 кодирует нетранслируемую РНК с неустановленной функцией. Этот ген тесно сцеплен с предыдущим геном IGF2; их экспрессия регулируется общим энхансером, расположенным в 3'-конце гена Н19, который имеет противоположный импринтинг с материнской хромосомы.
Тринадцатый ген L23 кодирует рибосомоподобный белок, экспрессирующийсябиаллельно в эмбриональных и зрелых тканях. Этим геном ограничена дистальная часть критической области 11р15.5.
К генетическим вариантам СБВ относятся:
• частичнаятрисомия дистальной части короткого плеча хромосомы 11 (результат аномальной сегрегации в мейозе структурных перестроек отцовского происхождения);
• спорадические дупликации критической области хромосомы 11 (11р15.5); их частота (совместно с частичной трисомией) - около 2%;
• однородительскаяотцовскаядисомия (результат постзиготической митотической нестабильности - рекомбинации с высокой долей мозаицизма);
|
• сбалансированнаятранслокация между хромосомами 11 и 22, унаследованная от матери.
Помимо этих генетических вариантов, идентифицированы точковые мутации в одном из двух генов-кандидатов СБВ - гене CDKN1C, c которым связывают 10-15% всех мутаций.
Другой ген-кандидат СБВ - IGF2 не имел точковых мутаций, но его экспрессия у больных проходила либо только с материнской хромосомы, либо наблюдалась ОРД по отцовской хромосоме 11, т.е. в данном случае имела место потеря импринтинга (ПИ).
Описан больной с унаследованной от матери сбалансированной инверсией короткого плеча хромосомы 11 с точками разрывов в области СБВ-ХР2, что также объяснялось ПИ при нормальном эпигенотипе Н19.
Таким образом, определенная часть спорадических случаев СВБ не связана с указанными выше генетическими вариантами, а объясняется ПИ. В общей сложности, структурные аномалии хромосомы 11 выявляются у 2% больных с СБВ. Первым по частоте повреждаемости является район локализации гена KvLQT1, обозначаемый как СБВ-ХР1; два других района - СБВ-ХР2 и СБВ-ХР3 - расположены ближе к центромере на 5 и 7 млнн.п. соответственно. В районе СБВ-ХР2 выделены два гена, содержащих мотив «цинковые пальцы». Это гены ZNF214 и ZNF215, второй ген имеет пять альтернативных и один антисмысловойтранскрипты, в норме экспрессирующиеся только с материнскогоаллеля. Структура хромосомного района СБВ-ХР3 пока не изучена.