

Болезни ошибок импринтинга Этиология
Болезни ошибок импринтинга - результат микроделеций в регуляторных областях импринтированных генов, или центрах импринтинга (ЦИ).
Первое сообщение о таких болезнях относится к 1993 г., когда у больных с СПВ и СА не было отмечено типичных делеций критического района хромосомы 15 (15q11.2-q13), но также (как и в случае ОРД) наблюдался одинаковый характер метилирования обеих родительских копий этого критического района. В дальнейшем выяснено, что в одном случае была одинаковой и соответствовала характеру метилирования на обеих родительских хромосомах экспрессия импринтированных генов, локализованных в критическом районе и характерных для СПВ (гены: SNRPN, ZNF127, PAR1, PAR5, IPW,
PWCR1, C15orf2).
Молекулярно-генетические исследования показали, что ошибки импринтинга часто сопровождаются микроделециями ЦИ. Сами делеции в ЦИ не связаны с клиническими проявлениями, но могут выявляться у здоровых родителей и других родственников больных пробандов. Анализ родословных в таких семьях свидетельствует, что делеции ЦИ наследуются (без клинических проявлений) в ряду поколений у лиц одного и того же пола. Таким образом, к заболеваниям этой группы относятся те же два синдрома (СПВ и СА), однако вызваны они другими генетическими причинами, т.е. это их генетические варианты.
|
Патогенез
При генетических вариантах СПВ и СА показано, что на одной из родительских хромосом изменен на обратный или инвертирован характер метилирования (и экспрессии) генов. Это объясняется переключением (эпигеномной заменой) генов хромосомы 15 в гаметогенезе одного из родителей, возникшим в результате ошибок в ЦИ. Предложены две модели такой замены в гаметогенезе:
• первая модель - эпигеномное переключение только в той гомологичной хромосоме, которая получена от родителя противоположного пола, тогда как вторую гомологичную хромосому модификация не затрагивает;
• вторая модель - это предварительное «стирание» существующей геномной «памяти» на обеих родительских хромосомах с последующим установлением «памяти», соответствующей данному полу; эта модель наиболее предпочтительна.
Отдельные нозологии Синдром Прадера-Вилли
СПВ впервые описан в 1956 г. Встречается с частотой 1:10-20 тыс. Как правило, возникает спорадически, хотя известны семейные формы.
Основные признаки: черепно-лицевой дисморфизм (долихоцефалия, миндалевидный разрез глазных щелей, гипертелоризм, эпикант, микрогнатия, «рыбий рот», высокое нёбо, диспластичные ушные раковины, акромикрия, нарушения дерматоглифики), низкий рост, ожирение, мышечная гипотония, гипогонадизм, гипопигментация, задержка умственного развития.
Продолжительность жизни больных не превышает 25-30 лет.
Этиология и механизмы патогенеза СПВ и «альтернативного» ему по генетической природе СА были изучены в последние годы. Выделены три генетические причины СПВ
Первая (основная) причина - это делеция критического района отцовской хромосомы 15 (15q11.2-q13) - 95% случаев генного импринтинга. Столь высокая частота этой мутации обусловлена мейотической нестабильностью указанного хромосомного района. Показано: делеции (как и в случае СА) могут возникать в гаметогенезе в результате внутриилимежхромосомных перестроек. Причем внутрихромосомныеделеции происходят путем образования большой петли, имеющей 3-4 млнн.п.
|
Рассмотрим молекулярную организацию критического района хромосомы 15. На рис. 1 приведена молекулярная структура этого критического района хромосомы 15.
На рис. 1 обозначены два кластера точек разрывов при делециях, картированныхпроксимальнее гена ZNF127, локализованного в этом критическом районе.
Здесь же расположены три белок-кодирующих гена (NDN, MAGEL2, SNRPN) и 7 генов, кодирущихнетранслируемые РНК (ZNF127AS, PAR5, PARSN, IPW, PAAR1, C15orf2, PWCR1), - все гены в этих локусах при СПВ экспрессируютсямоноаллельно на отцовской хромосоме. Для трех из указанных генов (гены NDN, ZNF127, SNRPN) показана тканеспецифическая экспрессия в нейронах головного мозга.
Неимпринтированная область хромосомы 15 находится в дистальной части критического района 15q11.2-q13. Она включает несколько биаллельно функционирующих генов, в том числе локус Р (отвечает за пигментацию кожи и глаз), гены GABRB3, GABRA3 и GABRG3 (кодируют субъединицы рецептора гамма-аминомасляной кислоты, или ГАМК-А), и ген HERC2. Последний ген филогенетически относится к самым древним генам: он считается родоначальником семейства END-псевдогенов, возникшего в результате нескольких последовательных дупликаций. Считается, что END-псевдогены способствуют процессам гомологической рекомбинации и неравного кроссинговера между повторяющимися последовательностями ДНК в критическом районе 15q11.2-q13.
К первопричинам СПВ, обусловливающим только 5% случаев генного импринтинга, относятся: инвертированная дупликация (тетрасомия), простая дупликация (трисомия), несбалансированная транслокация (моносомия), а также сбалансированная транслокация и инверсия критического района отцовской хромосомы 15 (15q11.2-q13).
Вторая причина СПВ - это ОРД по материнской хромосоме 15, обусловливающая 25% случаев болезни.
|
• Третья причина СПВ - это ошибки импринтинга. Генами-кандидатами СПВ являются следующие. Ген SNURF-SNRPN - состоит из 10 экзонов и имеет необычную бицистронную структуру. Его мРНК-транскрипт содержит две неперекрывающиеся рамки считывания для двух разных белков: первый белок SNURF (по-видимому, участвует в регуляции экспрессии импринтированных генов на отцовской хромосоме), второй белок N или SmN (является компонентом малых ядерных рибонуклеопротеинов - мяРНП). Анализ экспрессии двойного транскрипта гена SNURF-SNRPN показал его присутствие во всех тканях. Кроме двойного транскрипта, в мышечной и почечной тканях была обнаружена укороченная мРНК, соответствующая 1-34 экзонам этого гена.
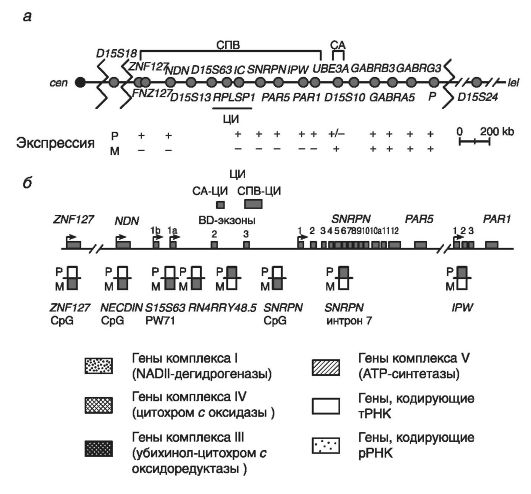
Рис. 1. [3] Молекулярная структура критического хромосомного района 15q11.2- q13 (по GenomicImprinting, 1997):
а - генетическая карта критического района 15(q11-q13), в которой положение генов обозначено кружками, точки разрывов хромосомы (при стандартных делециях) обозначены зигзагами; транскрипция отцовского (Р) и материнского (М) аллелей обозначена знаками: «+» - экспрессируется, «-» - не экспрессируется; б - характер аллельного метилирования локусов критического района 15 (q11-q13).
Экзоны генов показаны прямоугольниками, сайты инициации транскрипции каждого гена - стрелками. ЦИ имеет два альтернативных сайта инициации транскрипции. Отсутствие метилирования показано незакрашенными прямоугольниками, наличие метилирования - закрашенными прямоугольниками
• ZNF127 - в составе этого гена выделены две формы: собственно ZNF127 и антисенс ZNF127AS; обе формы транскрибируются в противоположных направлениях только на отцовской хромосоме. Ген содержит ряд мотивов типа «цинковые пальцы». Экспрессия гена ZNF127 показана во всех тканях с максимумом проявления в семенниках. Высокий уровень транскрипта ZNF127AS также показан в тканях мозга и легких; в других тканях его почти нет. Предполагается, что потеря экспрессии этих генов в ряде тканей организма обусловливает развитие таких симптомов, как бесплодие, гипогонадизм, неврологические и психические расстройства, ожирение.
|
• NDN - некдиновый ген. Он не содержит интронов и кодирует белок, угнетающий рост нейронов (подобно белку RBI при ретинобластоме, являющемуся супрессором опухолевого роста). Данный белок тормозит переход G1-cтадии клеточного цикла в стадию S. При этом некдин максимально экспрессируется в нейронах гипоталамуса, контролирующего процессы развития ожирения и гипогонадизма у больных.
• Локус Р - располагается в дистальной части критического района хромосомы 15, экспрессируетсябиаллельно, контролирует пигментацию кожи и радужки глаз. Потеря двух аллелей локуса Р ведет к альбинизму и гипопигментации кожи (у гетерозигот).