

Кривые титрования
Кривые титрования в аргентометрии обычно строят в координатах pAg — V, где pAg — взятый с обратным знаком логарифм концентрации ионов Ag+, V — объем титранта. В качестве титранта выступают AgNO3 или KSCN, может быть взят также раствор NaCl.
Рассчитаем кривую титрования 100,0 мл 0,10 М раствора NaCl раствором 0,10 М AgNO3, принимая для простоты расчетов, что объем раствора при титровании не изменяется и разница между активностью и концентрацией ионов несущественна.
В водном растворе NaCl диссоциирован нацело, поэтому концентрация хлорид-ионов С1~ в начальной точке кривой титрования, когда титрант еще не добавлялся, равна концентрации NaCl, т. е. 0,10 моль/л и рС1 = 1,00.
Добавление 1,00 мл раствора AgNO3 к NaCl вызовет образование осадка AgCl, и концентрация хлорид-ионов С1~ в растворе уменьшится. Равновесные концентрации ионов в растворе будут равны: [Ag+] = х, [С1~] = 0,099 + х. В соответствии с правилом произведения растворимости [Ag+] [С1~] = 1,78 • 10"10 можно записать: х(х + 0,099) = 1,78 • 10"10, откуда х = [Ag+] = = 1,78 • 10~™ = lg . 10-9 М0ЛЬ/Л; или pAg = 8,74. Добавление 90,0 мл раствора AgNO3 свяжет примерно 90% хлорида в осадок AgCl, и в растворе останутся равновесные концентрации [Ag+] = х-ц- = 1,78 • 10 6 моль/л, откуда pAg = 5,74. При добавлении 100,0 мл раствора AgNO3 достигается точка эквивалентности, в которой концентрации ионов Ag+ и СГ одинаковы:
[Ag+] = [СГ] = 71,78 • Ю-10 = 1,33 • Ю-5 моль/л и pAg = 4,88. Избыток в 0,10 мл раствора AgNO3 сверх эквивалентного приводит к концентрации [Ag+] = 10~4 + х и [СГ] = х, а так как х « 1,0 • 10"4, то [Ag+] = 1,0 • 10"4 моль/л и pAg = 4,0. Избы-, ток в 1,00 мл раствора AgNO3 дает [Ag+] = 1,0 • 10~3 моль/л и pAg =3,0. Результаты проделанных расчетов представлены графически на рис. 13.1. Кривая аргентометрического титрования, как показывает этот рисунок, сохраняет традиционный вид. Сначала от первых капель до оттитровывания 90% имеющегося хлорида pAg изменяется всего на одну единицу, затем то же изменение pAg вызывает только 9,0 мл раствора AgNO3, а в области точки эквивалентности наблюдается скачок титрования. Добавление 0,2 мл раствора AgNO3 в этой области (от 99,9 до 100,1 мл) вызывает изменение pAg от 5,74 до 4,00, т. е. почти на две единицы. Величина скачка титрования возрастает с увеличением концентрации реагирующих веществ и с уменьшением температуры, так как понижение температуры вызывает уменьшение ПР. Зависимость величины скачка титрования от ПР можно проследить, если рассчитать область скачка титрования для реакции взаимодействия AgNO3 с бромидом и иодидом натрия. Результаты расчетов также представлены на рис. 13.1, где наглядно видно увеличение скачка титрования с уменьшением ПР соли серебра, образующейся при титровании.
2. Индикаторы
В аргентометрии применяют различные способы установления точки эквивалентности как с помощью индикаторов, так и без них.
Метод равного помутнения. Идея метода предложена Гей-Люссаком. При титровании хлорида по этому методу вблизи точки эквивалентности отбирают небольшие порции прозрачного раствора и добавляют к одной порции AgNO3, а к другой NaCl. Если достигнута точка эквивалентности, жюмутнение в обеих порциях будет одинаковым. В недотитрованных растворах помутнение будет происходить только при добавлении AgNO3, а в перетитрованных — при добавлении NaCl. Метод равного помутнения характеризуется высокой точностью.
Метод Мора (индикатор — хромат калия). Идея метода основана на образовании кирпично-красного осадка хромата серебра Ag2Cr04 в точке конца титрования. При аргентометрическом титровании хлорида концентрация ионов в точке эквивалентности составляет [Ag+] = [С1~] = Vl,78 • Ю-10 = 1,33 х х 10~5 моль/л. Концентрация ионов CrOf~, необходимая для достижения ПРА СгО , при этой концентрации ионов [Ag+] однако при этой концентрации осадок хромата серебра «на глаз» не будет заметен. Для визуального обнаружения осадка хромата серебра обычно бывает достаточно перетитровать анализируемый раствор на одну каплю раствора AgNO3.
Титрование с хроматом в качестве индикатора проводится в нейтральной или слабощелочной среде, когда рН раствора больше 6,5, но меньше 10,5. В более кислой области происходит про-тонирование хромата (CrOf~" + Н+ = HCrOj) и чувствительность индикатора падает, а в более щелочных растворах, чем рН 10,5, оксид или гидроксид серебра может выпадать ранее хромата.
Метод Мора обычно применяют для определения хлоридов и бромидов. Иодиды и тиоцианаты не определяются, так как вследствие адсорбции установление точки эквивалентности становится затруднительным и погрешность анализа существенно возрастает.
Метод Фольгарда [индикатор — тиоцианатные комплексы же-леза(Ш)]. Реакцию взаимодействия серебра с тиоцианатом используют для определения галогенидов методом обратного титрования. По этому методу к анализируемому раствору галогенида (хлорида или бромида) добавляют избыток титрованного раствора AgNO3 и не
вошедшее в реакцию количество Ag+ оттитровывают тиоцианатом калия или аммония в присутствии ионов Fe3+ (метод Фольгарда).
Тиоцианатная реакция железа позволяет обнаружить в растворе тиоцианат при концентрации порядка 10~5 моль/л, что соответствует 0,01 мл 0,1 М KSCN в 100,0 мл раствора.
В точке эквивалентности концентрации ионов будут равны:
[Ag+] = [SCN-] = 7nPAgSCN = л/1,1 • Ю"12 = 1,05 • 10~6 моль/л. При этой концентрации тиоцианата окрашивание раствора не происходит. Первая же избыточная капля тиоцианата калия (аммония) вызывает четкую оранжевую окраску.
По методу Фольгарда могут быть оттитрованы и другие анионы, образующие малорастворимые соединения с ионом Ag+ (С2О|~, РО|" и т. д.). Существенным достоинством метода Фольгарда является возможность определения галогенидов в кислой среде.
Метод Фаянса (адсорбционные индикаторы). Адсорбционными индикаторами называют соединения, которые при адсорбции на осадке изменяют свой цвет. Установлено, что в первую очередь на осадке адсорбируются ионы, одноименные с осадком. Например, при титровании хлорида нитратом серебра на осадке AgCl до точки эквивалентности будут адсорбироваться преимущественно хлорид-ионы СГ~ и для нейтрализации отрицательного заряда к частицам осадка будут притягиваться положительно заряженные ионы из раствора. После точки эквивалентности адсорбироваться на осадке будут избыточные ионы Ag+ и для нейтрализации уже положительного заряда осадка из раствора будут притягиваться отрицательно заряженные ионы, в том числе анионы индикатора. Анионы некоторых красителей, адсорбируясь, изменяют свой цвет.
Отличным адсорбционным индикатором для титрования хлора является флуоресцеин, имеющий в растворе желто-зеленую окраску, а в точке эквивалентности окрашивающий осадок AgCl в красный цвет. Титрование с флуоресцеином происходит при рН 7—10, поскольку индикатор является слабой кислотой (р-йГ — 8). Дихлорфлуоресцеин уже более сильная кислота (трК — 4) и титрование хлорида с этим индикатором возможно при рН 4.
Эозин успешно применяется для титрования бромида, иоди-да и тиоцианата при рН 2, поскольку он является довольно сильной кислотой (рК 2). Известны также адсорбционные индикаторы-катионы, адсорбирующиеся на отрицательно заряженных осадках. Одним из таких индикаторов является родамин 6G. Титрование с адсорбционными индикаторами в оптимальных условиях характеризуется высокой точностью и надежностью.
Вопрос №18
Органические осадители. В количественном неорганическом анализе впервые применил органическое соединение М. А. Ильинский (1855—1941 гг.), предложивший в 1884 г. а-нитрозо-р-нафтол в качестве реагента на Co2+. Однако широкое использование органических реагентов началось после классических работ Л. А. Чу-гаева (1873—1922 гг.), предложившего в 1905 г. свою знаменитую реакцию на Ni2+ с диметилглиоксимом и выдвинувшего проблему изучения аналитических свойств внутрикомплексных солей. Работы Чугаева знаменовали начало нового, весьма плодотворного направления в аналитической химии, характеризующегося широчайшим использованием органических соединений в качестве реагентов на различные ионы. За протекший с тех пор период времени было открыто огромное число ценных органических соединений, применяемых ныне как в качественном, так и в количественном анализе. Основной причиной широкого проникновения органических реагентов в практику анализа является ряд особенностей их по сравнению с неорганическими реагентами.
1) Образующиеся соединения очень часто обладают весьма малой растворимостью в воде, что дает возможность при осаждении и промывании их избежать потерь вследствие растворимости.
2) Соосаждение сказывается в гораздо меньшей степени.
3) Содержание определяемого элемента в получающемся осадке оказывается более низким, так как органические осадители обладают обычно большим молекулярным весом. И когда осадок является весовой формой, фактор пересчета представляет величину сравнительно малую, что повышает точность определения.
4) Образующиеся продукты нередко интенсивно окрашены, что позволяет обнаруживать и колориметрически определять соответствующие ионы при ничтожно малых концентрациях их в растворе.
Реакции, происходящие при действии органических реагентов, относятся к различным типам. Из них особенный интерес для анализа представляет образование внутрикомплексных солей.
Применение органических осадителей требует создания определенных условий и прежде всего надлежащей величины рН раствора. Причину этого понять нетрудно. Выше указывалось, что при образовании внутрикомплексных солей происходит замещение водорода кислотной группы реагента ионами металла; при этом в раствор переходят ионы водорода, как это следует, например, из приведенного выше уравнения реакции между Ni2+ и ди-метилглиоксимом. Ясно, что положение равновесия должно зависеть от концентрации H+, т. е. от величины рН раствора. Диметнл-глиоксим (и другие подобные ему органические реагенты) ведет себя как слабая кислота. Поэтому к рассматриваемой реакции применимо все то, что говорилось ранее о значении величины рН при осаждении малорастворимых солей слабых кислот. И здесь, если известна величина ПР осадка и константа кислотной ионизации реагента, можно вычислить величину рН, при которой достигается полное осаждение.
При осаждении Ni2+ диметилглиоксимом, как и при других реакциях осаждения внутрикомплексных солей, происходит накопление H+ в растворе, и для смещения равновесия реакции вправо нужно эти ионы связывать. Следовательно, чем больше величина
рН раствора, тем полнее, казалось бы, должно быть осаждение. Однако в действительности приходится считаться с возможностью протекания различных побочных процессов, которые могут сделать слишком большое повышение рН невыгодным. Например, при этом могут выпадать в осадок соединения, образуемые данным реагентом с другими присутствующими в растворе катионами. В случае катионов, гидроокиси которых амфотерны, повышение рН вызовет превращение их в соответствующие анионы, например AlOjT, MoO4- и т. п., что может сделать невозможным осаждение соответствующей внутрикомплексной соли. Наконец, осаждение органическими реагентами часто проводится в присутствии различных маскирующих средств, например винной кислоты, действие которых также зависит от величины рН раствора.
Таким образом, при использовании органических осадителей ограничена не только нижняя, но и верхняя граница рН. Например, осаждать Ni2+ диметилглиоксимом следует при величинах рН, равных 5—10.
Органические осадители в большинстве случаев малорастворимы в воде. Поэтому их часто приходится применять в виде растворов в спирте, ацетоне и других неводных растворителях. Однако присутствие органических растворителей при осаждении повышает растворимость образующегося осадка и делает осаждение менее полным. Чтобы избежать этого, не следует прибавлять большого избытка осадителя.
Остановимся в заключение на кратком рассмотрении нескольких наиболее часто применяемых органических осадителей.
Диметилглиоксим. Структурная формула диметилглиокси-ма приведена выше. Это соединение применяется как важнейший реагент на Ni2+ для открытия и количественного определения его, а также для отделения от других катионов. Диметилглиоксим образует окрашенное в красный цвет, но растворимое комплексное соединение также с Fe2+, Pd2+ и некоторыми другими катионами.
8-0 кс и х и н о л и н C9H7ON, называемый часто о к с и н о м, имеет следующее строение:
8-Оксихинолин обладает амфотерным характером. Присутствие гидроксильной группы, связанной с бензольным ядром, обусловлю вает его кислотные свойства, а наличие третичного азота * —
* Третичным азотом называется атом азота, все три единицы валентности которого затрачены на- соединение с какими-либо атомами или радикалами, как это имеет место в третичных аминах, например „(CHa)3N, (CjHs)3N и т. п. Основные свойства оксихинолина проявляются в его способности к образованию солей с кислотами, например с CH3COOH, поэтому он гораздо лучше растворяется в присутствии CH3COOH, чем в чистой воде^
![]()
основные. Он осаждает большое число различных катионов, которые замещают атомы водорода гидроксильной группы и в то же время образуют с атомами азота координационную связь:
Здесь п (заряд катиона) показывает, что катион металла соединяется подобным образом не с одним, а с п однозарядными остатками 8-оксихинолина.
Потенциометрическое титрование широко используется в лабораторной практике. Оно применяется в тех случаях, когда надо провести экспресс-анализ вещества, а необходимых реактивов и оборудования нет или оно недостижимо в данное время. Создание новых моделей рН-метров, более компактных, надежных и удобных только повышает его востребованность.
Вопрос №40
. Понятие потенциометрического титрования;
Потенциометрический метод – это метод качественного и количественного анализа, основанный на измерении потенциалов, возникающих между испытуемым раствором и погруженным в него электродом. Данный метод рекомендуется для проведения анализов окрашенных растворов или малых концентраций веществ, для количественного анализа некоторых фармакопейных препаратов. Используя потенциометрическое титрование, можно более объективно устанавливать точку эквивалентности, поэтому метод находит широкое практическое применение, особенно в заводских лабораториях и экспресс-анализе. Помимо аналитических целей метод может быть использован для изучения кинетики химических процессов.
Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала индикаторного электрода. Это наблюдается, конечно, лишь тогда когда хотя бы один из участников реакции титрования является участником электродного процесса. Так, например, титрование по методу кислотно-основного взаимодействия может быть выполнено со стеклянным электродом. Определение хлорида - с хлорсеребряным и т.д. Так же, как и в других титриметрических методах, реакции потенциометрического титрования должны протекать строго стехиометрически, иметь высокую скорость и идти до конца.
Главной особенностью потенциометрического титрования есть использование различных видов электродов от выбора которых напрямую зависит точность проведенных исследований. Поэтому при проведении титрования надо учитывать некоторые особенности электродов:
Если есть возможность выбора – предпочтение следует отдавать электродам с наименьшим электрическим сопротивлением, т.к. это позволит снизить электростатические наводки и сделать измерения более точными, быстрыми и комфортными;
При анализе щелочных растворов с высоким содержанием ионов натрия следует применять высокоомные электроды;
Для анализа растворов имеющих повышенную температуру (>50°С) предпочтительны высокоомные электроды, т.к. в этих условиях их сопротивление значительно снижается, и они приобретают все положительные свойства низкоомных электродов, но при этом имеют более широкий диапазон измерений и больший ресурс работы.
Виды потенциометрического титрования;
Классификация потенциометрических методов анализа такова, как и обычного объемного анализа. В ее основу положены типы химических реакций: нейтрализации, осаждения, комплексобразования, окисления – восстановления и т. п.
Кислотно-основное титрование используют для нахождения концентраций сильных кислот и оснований, слабых кислот и их солей во всех случаях, когда использование цветных индикатор затруднено.
Принцип метода осаждения и комплексообразования состоит в получении исследуемых ионов в виде нерастворимых веществ или в виде стойких растворимых комплексных соединений. В этом случае при титровании изменяется концентрация иона металла в растворе. Как индикаторные используют серебряный и ртутный электроды.
Для комплексонометрических титрований может быть использован универсальный электрод Hg|HgY2- или Au(Hg)|HgY2- где Au(Hg) - амальгамированное золото; HgY2- - комплекс ртути с анионом этилендиаминтетрауксусной кислоты. С помощью ртутного электрода этого типа могут быть оттитрованы любые ионы, которые образуют с Y4- комплексы с константой устойчивости, не превышающей константу устойчивости ртутного комплекса. Это, например, ионы магния Mg2+, кальция Ca2+, кобальта Co2+, никеля Ni2+, меди Cu2+, цинка Zn2+ и др.
Индикаторными электродами в методах потенциометрического титрования, использующих реакции осаждения, служат металлические или мембранные электроды, чувствительные к определяемому иону или иону-осадителю. Практически по методу осаждения могут быть определены катионы серебра, ртути, цинка, свинца, анионы хлора, брома, иода и некоторые другие. Смесь галогенидов, например I- и Cl-, может быть оттитрована без разделения нитратом серебра. Серебряный электрод позволяет фиксировать два скачка в ходе такого титрования. Первый скачок свидетельствует об оттитровывании иодид-иона и может быть использован для расчета содержания этого иона, второй скачок относится к окончанию осаждения хлорид-иона. По второму скачку можно рассчитать суммарное содержание галогенидов или концентрацию хлорид-иона, если концентрация иодид-иона будет известна из данных по титрованию до первого скачка.
Вопрос №28
При окислительно-восстановительном титровании индикаторными электродами будут индиффирентные металлы: платина. Палладий и золото. Наиболее часто т в потенциометрии используют гладкий платиновый электрод. Переход потенциала индикаторного электрода от одной окислительно-восстановительной системы к другой сопровождается скачком потенциала и свидетельствует об окончании процесса титрования.
Кривые окислительно-восстановительного титрования могут быть построены в координатах или pM – V (титранта) или E – V (титранта), E – потенциал системы, V (титранта) – объем титранта. Кривые титрования первого типа представляют практический интерес, когда имеется индикаторный электрод, чувствительный к данному веществу. Кривые второго типа имеют более общее значение, так как любое окислительно-восстановительное титрование может быть проведено по измерению E с использованием индикаторного электрода из благородного металла.
Вопрос №24.
Кислотно-основное титрование.
Кислотно-основное титрование получило также название ацидиметрии и алкалиметрии. В основе этих методов лежат протолитические реакции, в результате которых происходит связывание ионов Н3О+ и ОН- в воду:
![]()
В кислотно-основном титровании в качестве индикаторного обычно используют стеклянный электрод, как правило, входящий в комплект серийно выпускаемых промышленностью pH-метров. Потенциометрический метод позволяет провести количественное определение компонентов в смеси кислот, если константы диссоциации различаются не менее чем на три порядка. Например, при титровании смеси, содержащей хлороводородную (HCl) и уксусную кислоты, на кривой титрования обнаруживается два скачка. Первый свидетельствует об окончании титрования HCl, второй скачок наблюдается при титровании уксусной кислоты. Также несколько скачков имеют кривые титрования многоосновных кислот, константы диссоциации которых существенно различаются (хромовая, фосфорная и др.).
Ниже мы детально рассмотрели теоретические кривые для различных случаев кислотно-основного титрования. Форма этих кривых может быть близка к форме экспериментальных кривых. Часто, однако, экспериментальные кривые смещены относительно теоретических, поскольку при построении последних, обычно, оперируют концентрациями, а не активностями. Изучение теоретических кривых показывает, что небольшая погрешность потенциометрического определения рН не имеет значения при определении конечной точки титрования.
Потенциометрическое кислотно-основное титрование особенно удобно при анализе смесей кислот или многоосновных кислот (оснований), поскольку оно часто позволяет достичь разделения конечных точек титрования. Из кривых потенциометрического титрования можно также определить приближенные значения констант диссоциации реагирующих веществ. Теоретически эту величину можно рассчитать из любой точки на кривой титрования, практически же ее легче найти из значения рН в точке полунейтрализации. Например, при титровании слабой кислоты НА в средней точке можно предположить, что
![]()
и поэтому
![]()
рКа = рН .
Важно заметить, что константы диссоциации, определенные этим способом, отличаются от констант, приводимых в таблицах, поскольку последние включают активности, в то время как первые – концентрации. Так, если мы запишем выражение для константы диссоциации в более точном виде, то получим
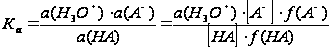
Примем, что , поэтому
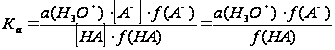
Прологарифмировав обе части уравнения и изменив знак, получим, что:
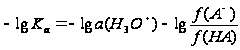
Затем после приведения к р-функции и преобразования получаем:
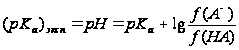
Таким образом, истинное значение рКа
будет отличаться от экспериментального
на величину логарифма отношения
коэффициентов активности. Обычно при
титровании ионная сила равна 0,1 или
больше, следовательно, отношение
![]() должно
быть по крайней мере 0,75, если НА не
заряжена. Для таких соединений, как
должно
быть по крайней мере 0,75, если НА не
заряжена. Для таких соединений, как
![]() или
или
![]() это
отношение должно
это
отношение должно
быть даже выше.
Из одного-единственного потенциометрического титрования можно получить и величину эквивалентной массы, и приблизительное значение константы диссоциации очищенной пробы неизвестной кислоты; часто эта информация достаточна для идентификации кислоты.
Вопрос №23
Определение конечной точки титрования.
Для определения конечной точки потенциометрического титрования можно использовать различные способы. Наиболее простой состоит в построении графика зависимости потенциала или рН от объема реагента (рис. 2.1. а). Затем визуально определяют среднюю точку участка, соответствующего вертикальному подъему кривой, и принимают ее за конечную точку. Предложены различные механические способы установления средней точки, но они ненамного улучшают точность ее нахождения.
Второй способ состоит в расчете изменения потенциала на единицу изменения объема реагента (т. е. нахождение ΔЕ/ΔV). График, построенный с использованием этого параметра как функции относительно объема, имеет острый максимум в конечной точке (рис. 2.1. б). С другой стороны, это отношение можно вычислить во время титрования и записать вместо потенциала. Как следует из данных, максимум находится между 24,3 и 24,4 мл; выбор 24,35 мл будет пригоден для большинства случаев.

Кривая потенциометрического титрования.
а) титрование 2,433 мэкв СІ- 0,1 н рас твором AgCI; б) кривая по первой производной; в) кривая по второй производной.
Лингейн [6] показал, что объем реагента можно зафиксировать более точно, определив точку, в которой вторая производная потенциала по объему (т. е. Δ2Е/ΔV2) равна нулю. Эти вычисления несложны, если вблизи точки эквивалентности добавляют равные порции раствора реагента.
Эта функция должна обратиться в нуль в некоторой точке между двумя объемами, где происходит перемена знака. Соответствующий этой точке объем получают интерполированием. Кривая в на рис. 2.1 представляет собой график зависимости Δ2Е/ΔV2.
Рассмотренные выше способы основаны на предположении, что кривая титрования симметрична относительно точки эквивалентности и перегиб кривой соответствует этой точке. Это допущение совершенно справедливо при условии, что вещества, участвующие в химической реакции, взаимодействуют друг с другом в эквимолярных соотношениях и что электродный процесс полностью обратим. Если эти условия не выполняются, получается асимметричная кривая титрования. Отметим, что кривая титрования железа (II) раствором церия (IV) симметрична относительно точки эквивалентности. С другой стороны, каждый моль перманганата окисляет пять молей железа (II), что приводит к получению совершенно асимметричной кривой титрования. Обычно вблизи точки эквивалентности этих кривых изменения потенциала достаточно велики, и поэтому если за конечную точку принять среднюю точку круто восходящего участка кривой титрования, то ошибка титрования будет незначительна. Только в том случае, если требуется чрезвычайно высокая точность или, если работают с очень разбавленными растворами, следует учитывать этот источник ошибок. При необходимости можно ввести эмпирическую поправку, проведя титрование стандартного раствора. С другой стороны, когда ошибка обусловлена несимметричностью реакции, точное положение точки эквивалентности можно рассчитать теоретически [6].
Другие способы обнаружения конечной точки включают титрование до теоретически рассчитанного значения потенциала электрода или, лучше, до потенциала, эмпирически установленного
Вопрос №39
Кондуктометрические методы анализа основаны на измерении электропроводности исследуемых растворов. Существует несколько методов кондуктометрического анализа:
прямая кондуктометрия – метод, позволяющий непосредственно определять концентрацию электролита путем измерения электропроводности раствора с известным качественным составом;
кондуктометрическое титрование – метод анализа, основанный на определении содержания вещества по излому кривой титрования. Кривую строят по измерениям удельной электропроводности анализируемого раствора, меняющейся в результате химических реакций в процессе титрования;
хронокондуктометрическое титрование – основано на определении содержания вещества по затраченному на титрование времени, автоматически фиксируемого на диаграммной ленте регистратора кривой титрования.
Кондуктометрия
Кондуктометрия относится к наиболее распространенным методам исследования растворов и жидких систем вообще.
проводящими принято условно с χ ~10-7 Ом-1·см-1 и выше;
умеренно проводящими с χ: 10-7 – 10-11 Ом-1 ·м-1;
непроводящими – χ ниже 10-11 Ом-1 ·м-1.
Данная классификация условна.
В ФХА принято пользоваться диаграммами «удельная электропроводность χ – состав». Поскольку электропроводность относится к заведомо не аддитивным свойствам, способ выражения концентрации при этом может быть произвольным, однако для наглядности чаще всего выбирают мольные доли. Диаграммы «молекулярная электропроводность λ – состав» используется реже.
Электрическое сопротивление
Основной константой, характеризующей электрические свойства вещества, является удельное электрическое сопротивление, зависящее от природы вещества и от температуры.
Кондуктометрия располагает несколькими законами:
1. В очень разбавленных растворах (предельно разбавленных) эквивалентная электропроводность (λ0) является постоянной характеристикой раствора, не зависящей от изменения концентрации электролита. Говоря простым языком, это означает, что в разбавленных растворах электропроводность прямо пропорциональна количеству заряженных частиц – ионов.
Для растворов сильных электролитов область предельного разбавления простирается до концентрации 0,0001н, а с небольшой погрешностью можно считать границей предельного разбавления концентрацию 0,001н.
Для расчетов в области больших концентраций существует формула Кольрауша, но ее нельзя использовать для прогноза, поскольку она носит явно эмпирический характер:
λ = λ0 + K C 1/2,
2. Предельная эквивалентная электропроводность раствора электролита равна сумме эквивалентных электропроводностей катиона и аниона.
3. Эквивалентные электропроводности подавляющего числа ионов близки друг к другу по величине. Анализ экспериментальных данных показывает, что при 18 0С для катионов λ0=0,0053 ± 0,0019 Ом-1 м2 г-экв-1 и для анионов λ0=0,0055 ± 0,0027 Ом-1 м2 г-экв-1. При 25 0С λ 0=0,0062 ± 0,0023 Ом-1 м2 г-экв-1 для катионов и λ0=0,0064 ± 0,0031 для анионов. Исключение составляют ионы H+, OH-, Fe(CN)63-, Fe(CN)64-, электропроводности которых аномально высоки: