

Мышечные дистрофии
Эти дистрофии — разнородная группа наследственных заболеваний, которые клинически характеризуются выраженной слабостью и истощением мышц и развиваются в детстве.
ЛГ-связанные мышечные дистрофии. Две наиболее распространенные формы мышечной дистрофии имеют в своей основе аберрации, связанные с хромосомой X (см. главу 8): мышечная дистрофия Дюшенна (G.B.A. Duchenne) и мышечная дистрофия Беккера (P.Е.Becker).
Мышечная дистрофия Дюшенна. Она является наиболее тяжелой разновидностью указанных генетических нарушений, встречается примерно у 1 из каждых 10 ООО мальчиков. Выраженная форма мышечной дистрофии Дюшенна про- является к концу первых 5 лет жизни сильной мышечной слабостью, которая к 10—12 годам приводит к необходимости пользоваться креслом-коляской. Эта слабость прогрессирует до наступления смерти больного в возрасте примерно 20 лет. Мальчики, больные мышечной дистрофией Дюшенна, рождаются нормальными, ранние двигательные функции развиваются у них вовремя. Однако у таких больных нередко задерживается развитие способности к ходьбе, и первыми симптомами мышечной слабости являются неуклюжесть и отставание в развитии от сверстников. Слабость начинается с мышц тазового пояса, затем распространяется на плечевой пояс.
Важным клиническим признаком служит увеличение икроножных мышц, связанное со слабостью и называемое псевдогипертрофией. Увеличение мышечного объема первоначально обусловлено утолщением мышечных волокон, а позднее, по мере атрофии мышц, возрастанием объема жира и соединительной ткани. Патологические изменения отмечают также в сердце, у больных может развиться сердечная недостаточность или аритмия. Хотя в головном мозге нет четко выраженных структурных изменений, все же при мышечной дистрофии Дюшенна нарушаются познавательные способности. Причем у некоторых пациентов это настолько выражено, что может идти речь об умственной отсталости. В первые 10 лет жизни у пациентов повышен уровень сывороточной креатининкиназы, который, однако, возвращается к норме на более поздних стадиях заболевания. Смерть наступает вследствие дыхательной или сердечной недостаточности, а также легочной инфекции.
Примерно у 30 % больных мышечной дистрофией Дюшенна обнаруживают новые мутации. В остальных случаях женщины, являющиеся в соответствующих семьях обязательными носителями генетического дефекта, обычно не имеют никаких клинических симптомов. Правда, у них повышен уровень креатининкиназы, а в биоптатах из скелетной мускулатуры выявляют минимально выраженные гистологические признаки миопатии. В последние годы получено множество молекулярно-генетических и биохимических данных, объясняющих некоторые стороны патогенеза и диагностики мышечной дистрофии Дюшенна. Ген, ответственный за эту аномалию, имеет громадные размеры (2,5х106 пар оснований, содержащих более 80 экзонов). Он локализуется в районе Хр21, кодирующем белок с массой 427 kD, который называется дистрофином. Огромный размер гена значительно затрудняет генетический анализ, но все же стало известно, что большая часть мутаций представлена делециями, а остальная часть аберраций обеспечивается сдвигами рамки считывания и точковыми мутациями (см. главу 8). С помощью им- муногистохимических исследований (с применением окрашенных антисывороток к дистрофину), а также использования
полимеразной цепной реакции показано, что в нормальных мышечных волокнах дистрофии обычно прилежит к сарколемме. Вместе с тем в биоптатах, полученных из мышц больных, найдено минимальное количество дистрофина.
Мышечная дистрофия Беккера. Она обусловлена тем же генетическим локусом, что и предыдущее заболевание. Однако эта дистрофия встречается реже и протекает легче, чем мышечная дистрофия Дюшенна. Болезнь наступает в более позднем периоде детства или подростковом возрасте. Она развивается медленнее, но с различным темпом прогрессирования. У многих пациентов почти нормальная продолжительность жизни. Поражения сердца встречаются редко. Хотя взаимосвязь между мышечными дистрофиями Дюшенна и Беккера на основании клинических проявлений предполагалась давно, аллельный характер аберраций при этих двух заболеваниях установлен в настоящее время с несомненностью. При мышечной дистрофии Беккера тоже обнаружены мутации гена дистрофина. У больных выявлен белок с аномальной молекулярной массой, что отражает наличие мутаций, приводящих к синтезу дистрофина. Молекула указанного белка локализуется в сайтах полос / и М (см. главу 8). Полагают, что она играет определенную роль в поддержании непрерывности сарколеммы во время изменений формы мышечного волокна при его сокращениях.
Гистологические изменения, общие для мышечных дистрофий Дюшенна и Беккера, следующие: изменения диаметра мышечных волокон (из-за наличия как маленьких, так и гигантских, иногда расщепленных, волокон); увеличение количества внутренних ядер (выше нормального уровня 3 %); дегенерация, некроз и фагоцитоз мышечных волокон; регенерация мышечных волокон; пролиферация соединительной ткани эндомизия (рис. 24.19). При мышечной дистрофии Дюшенна также нередко определяются увеличенные, закругленные и гиалинизированные волокна, утратившие свою нормальную поперечную исчерчен- ность. Их считают слишком сильно сократившимися волокнами. Такие находки редки при мышечной дистрофии Беккера. У больных обоими видами мышечной дистрофии поражены волокна обоих типов, причем разница в распределении этих типов в пораженной мышечной ткани не выявлена. При мышечной дистрофии Дюшенна не всегда удается определить типы волокон с помощью гистохимических реакций. На более поздних стадиях мышцы постепенно и почти полностью замещаются жиром и соединительной тканью, приобретая вид, который принципиально не отличается от такового на конечной стадии других тяжелых мышечных поражений. Если в процесс вовлекается сердечная мышца, то в миокарде развивается интерстициальный фиброз, имеющий более выраженный характер в субэн- докардиальных слоях. Несмотря на клинические признаки нарушения функции головного мозга, какие-либо определенные
Рис.
24.19. Прогрессирующая мышечная дистрофия
на ранней стадии развития. Мышечные
волокна окружены и разделены фиброзной
тканью. В центре имеется скопление
макрофагов, поглощающих разрушенное
мышечное волокно [из Grundmann
Е.,
Geller
S.A., 1989].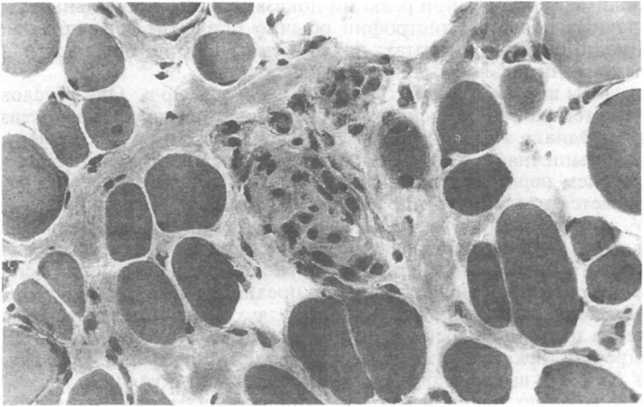
нейропатологические аномалии при мышечных дистрофиях не описаны.
Миотоническая дистрофия. Миотония — патологическое состояние мышц, выражающееся в их непроизвольном сокращении и устойчивом затруднении расслабления. Это основной нейромы- шечный симптом указанного заболевания. Больные часто жалуются на тугоподвижность и трудности при разжатии мышц, например при рукопожатиях. Часто приступ миотонии может быть вызван с помощью поколачивания по возвышению большого пальца. Заболевание начинается в позднем периоде детства с затруднений походки, вторичных по отношению к слабости задних сгибателей стоп. Затем слабость распространяется на глубокие мышцы кистей и разгибатели запястьев. Позднее развиваются атрофия мышц лица и птоз (опущение верхнего века, обусловленное поражением мышцы, поднимающей веко). Это приводит к характерному внешнему виду лица. Практически у каждого больного мио- тонической дистрофией выявляют катаракту (частичное или полное помутнение хрусталиков глаз со снижением остроты зрения вплоть до полной его утраты). Другие сопутствующие изменения'. преждевременное облысение, атрофия половых желез, кар- диомиопатия, вовлечение в процесс гладкой мускулатуры, снижение плазменного содержания IgG и аномальный результат теста на устойчивость к глюкозе. Изредка развивается слабоумие.
Поскольку заболевание наследуется по аутосомно-доми- нантному типу, оно имеет тенденцию к более тяжелому течению и более раннему проявлению в последующих поколениях. Такой феномен называют антиципацией. Миотоническая дистрофия может проявляться в виде врожденной слабости, и тогда она связана с наследственностью по материнской линии. Болезнь отличается тяжелой симптоматикой, врожденной лицевой диплегией (двусторонним параличом мимической мускулатуры, обусловленным гипоплазией клеток ядра лицевого нерва), трудностями при еде и дыхательной недостаточностью.
Ген, обусловливающий развитие миотонической дистрофии, локализуется на хромосоме 19ql3.2=13.3. Клонирование показало, что ген кодирует протеиновую киназу, которая называется ми- отонинпротеинкиназой. Локализованная на З'-нетранслированной области гена, она является тринуклеиновой дупликацией, состоящей из (CNG)n. Фенотип заболевания связан с расширением этой области; у здоровых людей имеется меньше, чем 30 дупликаций, в то время как у больных их может быть несколько тысяч. Мутация не стабильна в одном поколении; с каждым поколением накапливается больше дупликаций, что соответствует феномену антиципации. Расширение тринуклидных дупликаций влияет на конечный уровень протеинового продукта.
При патоморфологическом исследовании в скелетных мышцах могут обнаруживаться черты дистрофии, схожей с мышечной дистрофией Дюшенна (см. выше). Кроме того, значительно возрастает количество внутренних ядер, которые в продольном гистологическом срезе могут формировать целые цепочки. Другим отличительным признаком является наличие кольцевидных волокон с суб- сарколеммным ободком цитоплазмы, отчетливо определяемым с центральной стороны этих волокон. Указанный ободок содержит миофибриллы, которые ориентированы по круговому типу по сравнению с продольно ориентированными миофибриллами в остальной части волокна. Кольцевидное волокно может быть обусловлено неравномерной субстанцией саркоплазмы (саркоплазма- тической массой), простирающейся кнаружи от этого волокна. При окраске гистологических срезов гематоксилином и эозином кольцевидные волокна приобретают голубой цвет, а при окраске срезов по трихромовой методике Гомори (G.Gomori) — красный цвет, наконец, при гистохимической реакции с восстановленным никотинамидадениндинуклеотидом (NADP) — синий цвет. Связь между наличием кольцевидных волокон и клиническими проявлениями миотонии неясна. У некоторых лиц на ранних этапах болезни с помощью гистохимических исследований обнаружена относительная атрофия волокон I типа. Из всех мышечных дистрофий только миотоническая дистрофия сопровождается патологическими изменениями в интрафузальных волокнах (находящихся внутри мышечных веретен) с их расщеплением, некрозом и последующим восстановлением.