

Опухоли у детей
Опухоли у детей встречаются гораздо реже, чем у взрослых. Злокачественные новообразования детского возраста составляют всего 2 % от всех злокачественных опухолей человека. Однако среди причин смерти у детей они занимают одно из ведущих мест. В экономически развитых странах смертность детей от злокачественных новообразований занимает второе место после несчастных случаев и составляет 10 %. Опухоли детского возраста имеют ряд особенностей, значительно отличающих их от опухолей взрослых. Часто они возникают из остатков эмбриональных тканей в результате нарушения формирования органов и тканей в периоде внутриутробного развития. Такие опухоли называют дизонтогенетическими. У взрослых дизонтогенетичес- кие новообразования встречаются редко, тогда как у детей это преобладающий вид опухолей. До 85 % злокачественных опухолей детей в возрасте до 1 года представлены дизонтогенетическими опухолями. Довольно часто выявляют связь между ростом опухоли (онкогенезом) и пороками развития (тератогенезом). Так, опухоль Вильмса и гепатобластома часто сочетаются с гемигипертрофией (увеличением размеров одной половины туловища, конечностей или лица). Опухоли центральной нервной системы комбинируются с пороками развития мозга, опухоли гонад часто встречаются одновременно с пороками половых органов. Суммарно у 30 % детей с опухолями различной этиологии диагностируют пороки развития.
Отмечают большую роль генетических факторов в развитии опухолей детского возраста. Известно более 100 наследственных синдромов, которые предрасполагают к развитию опухоли в детском возрасте [по Cotran R.S., Kumar V., Collins Т., 1998]. Доказано, что в этиологии ряда врожденных опухолей генетические факторы имеют основное значение. Наследственный характер установлен для ретинобластомы, нефробластомы, ней- робластомы.
Доброкачественные опухоли бывают у детей гораздо чаще, чем злокачественные. Они составляют более 80 % новообразований у детей до 14 лет. Среди злокачественных опухолей преобладают опухоли кроветворной ткани, центральной нервной системы и саркомы.
|
Опухоли Частота (% от всех детских опухолей) Лейкозы |
30 |
Лимфомы, в том числе |
14 |
лимфогранулематоз |
|
Нейробластома |
6,8 |
Рабдомиосаркома |
6,5 |
Опухоль Вильмса |
5,2 |
Медуллобластома |
5 |
Ретинобластома |
2,7 |
Г епатобластома |
0,9 |
Рак встречается относительно редко (не более чем в 6 % случаев), в то время как у взрослых лиц карциномы весьма распространены. Критерии, принятые в патологии для характеристики злокачественных и доброкачественных опухолей (см. главу 7), не всегда бывают применимы к детским новообразованиям. Так, резко выраженный клеточный атипизм и полиморфизм могут наблюдаться у детей при некоторых доброкачественных опухолях надпочечников. Многие доброкачественные новообразования у маленьких детей могут расти очень быстро (например, невусы, гемангиомы). В таких случаях, несмотря на доброкачественное строение узла, можно найти большое число фигур митоза. В то же время инфильтрирующий рост типичен для ювенильной фибромы, капиллярной гемангиомы, лимфангио- мы. Напротив, некоторые злокачественные опухоли в первые годы жизни ребенка растут очень медленно. Нефробластома и нейробластома обычно имеют тонкую капсулу и некоторое время растут в ее пределах. Отмечена уникальная способность некоторых новообразований у детей к «дозреванию»: нейроблас- тома может превратиться в ганглионейрому, злокачественная гепатобластома в доброкачественную аденому, тератобластома в тератому. Это совершенно необыкновенное явление, не согласующееся с прогрессией злокачественных опухолей (см. главу 7), до конца не объяснено. Оно наблюдается в тех новообразованиях, которые возникают либо из эмбриональных тканей, задержавшихся в развитии по сравнению с другими тканями ребенка, либо из стволовых недифференцированных (камбиальных) клеток.
Опухоли у детей имеют особенности метастазирования. Саркомы мягких тканей у детей в 30 или 50 % случаев метастазиру- ют по лимфатическим сосудам. Это тоже не согласуется с представлениями о преимущественно гематогенном метастазирова- нии сарком. Напротив, эмбриональные гепатобластомы, т.е. эпителиальные опухоли, дают первые метастазы не в региональные лимфатические узлы, а в легкие. Большинство новообразований центральной нервной системы вообще не метастазирует за пределы черепа. В целом прогноз при злокачественных опухолях у детей, как правило, более благоприятный, чем у взрослых лиц. Все это заставляет уделять больше внимания уменьшению неблагоприятных последствий и отдаленных эффектов от химио- и радиотерапии у выживших детей (включая развитие вторичных опухолей и генетических последствий).
При рубрифицировании опухолей у детей не всегда можно применить гистогенетический принцип, принятый для новообразований у взрослых, так как дизонтогенетические опухоли могут состоять из элементов разных зародышевых листков. В зависимости от происхождения опухоли у детей подразделяют на три типа: дизонтогенетические, растущие из камбиальных эмбриональных тканей и развивающиеся по типу опухолей взрослых.
От истинных новообразований следует отличать опухолеподобные состояния — тератомы, гамартомы и хористомы (см. главу 7).
Доброкачественные опухоли. Наиболее частые доброкачественные опухоли у детей — гемангиомы, лимфангиомы, фибромы и тератомы. Строение этих опухолей подробно описано в других главах; здесь мы приведем особенности этих опухолей у детей.
Гемангиома. Это наиболее частая доброкачественная опухоль детского возраста. Обычно у детей встречаются капиллярная и кавернозная формы (см. главу 11) или их комбинация. Гемангиома локализуется в основном в коже головы, шеи или туловища, реже во внутренних органах. Капиллярные гемангиомы могут увеличиваться в размерах, особенно быстрый рост наблюдается в первые месяцы жизни. При возрасте ребенка 1—3 года рост опухоли останавливается, а к 5 годам она обычно подвергается спонтанной регрессии. Иногда гемангиома обладает инфильтрирующим ростом, в связи с чем возможны рецидивы. Гигантская гемангиома конечностей и туловища у детей грудного возраста может сопровождаться развитием тромбоцитопе- нической пурпуры вследствие распространенного тромбоза сосудов гемангиомы (синдром Казабаха—Мерритта, H.H.Kasa- bach, К.К.Merritt). Гемангиомы являются одним из проявлений наследственного синдрома Хиппеля—Линдау (Е. von Hippel, A.Lindau).
Лимфангиома. Обычно она выявляется у детей в возрасте до 3 лет. Опухоль имеет наибольшее клиническое значение, когда локализуется в глубоких областях шеи, подмышечной впадине, средостении и забрюшинном пространстве. Несмотря на отсутствие клеточного атипизма, лимфангиомы обладают местнодеструирующим ростом и увеличиваются в размерах после рождения. В этой связи они могут повреждать жизненно важные органы (например, в средостении) или нервные стволы. Лимфангиомы следует отличать от лимфангиэктазий — аномально расширенных предсуществующих лимфатических сосудов. Лимфангиэктазии сопровождаются диффузным отеком конечности или ее части, вызывая ее деформацию. В отличие от лимфангиом лимфангиэктазии не прогрессируют, однако могут вызывать серьезные косметические проблемы.
Фиброзные опухоли. У детей они разнообразны и часто вызывают большие трудности при определении степени злокачественности. Фиброматозы, встречающиеся у детей, нередко характеризуются гиперцеллюлярностыо и быстрым инфильтрирующим ростом, что делает их трудно отличимыми от фибросарком взрослых. Наблюдаются случаи спонтанной регрессии. Из множества фиброзных опухолей приведем для примера лишь некоторые. Инфантильный миофиброматоз характеризуется наличием небольших плотных узелков в дерме, подкожной клетчатке, мышцах, внутренних органах. Узелки могут быть одиночными и множественными (более 50). При одиночных опухолях прогноз благоприятный, при множественных — дети умирают на 1-м году жизни. Ювенильная ангиофиброма носоглотки встречается обычно у мальчиков старше 8 лет. Микроскопически опухоль состоит из полей фиброзной ткани с небольшим количеством фибробластов и тонкостенными кровеносными сосудами. Обладает инфильтрирующим ростом и иногда прорастает кости черепа. Локализация опухоли и характер ее роста затрудняют радикальное удаление. При повреждении или попытке удаления может развиться профузное кровотечение. Опухоль гистологически доброкачественная, метастазов не дает. Может подвергаться спонтанной регрессии.
Тератома. Это опухоль из эмбриональных недифференцированных половых клеток возникает при нарушении их миграции в период морфогенеза половых желез зародыша (см. главу 7). Тератома представлена тканями трех зародышевых листков, однако основную часть обычно составляют ткани эктодермального происхождения. В зрелой тератоме опрелеттяют- ся эпидермис со всеми производными (волосы, железы), глиальная ткань, скопления ганглиозных клеток, жировая и мышечная ткани, хрящ, реже — другие ткани в самых разных сочетаниях. Могут быть также элементы трофобласта. Тератомы имеют следующую наиболее типичную локализацию: яичники и яички, крестцово-копчиковая область, средостение, забрюшин- ное пространство, зев, основание черепа. У новорожденных и детей первых 2 лет жизни преобладают крестцово-копчиковые тератомы, с 15—16-летнего возраста увеличивается чягтотя яичниковых тератом. Бблыиая часть тератом яичка появляется в возрасте 20—49 лет. Крестцово-копчиковая тератома — основная разновидность тератом новорожденных и детей младшего возраста. У девочек она встречается в 3 раза чаще, чем у мальчиков, часто сочетается с неиммунной водянкой плода, многово- дием. Может вызывать затруднения при родоразрешении. В основном крестцово-копчиковые тератомы состоят из зрелых тканей, чаще с органоидной дифференцировкой. Злокачественные тератомы (тератобластомы) содержат элементы крупноклеточной карциномы, формирующей сосочковые структуры или растущей солидно. Для тератобластом характерно присутствие элементов опухоли эндодермального синуса. Такие опухоли имеют очень неблагоприятный прогноз.
Злокачественные опухоли. Наиболее часто злокачественные опухоли у детей развиваются в кроветворной системе и нервной ткани, мягких тканях, костях, почках. Частота распределения опухолей по органам резко контрастирует с аналогичным показателем у взрослых. У последних чаще поражаются легкие, молочная железа, кожа, предстательная железа, толстая кишка (см. главу 7). Заболеваемость злокачественными опухолями значительно варьирует в зависимости от возраста ребенка.
Злокачественные опухоли у детей имеют, как правило, ди- зонтогенетическое происхождение. Под микроскопом выявляют больше примитивных (эмбриональных), чем плеоморфно-ана- пластических признаков, что часто напоминает органогенез, специфический для органа, в котором развилась опухоль. Такие новообразования нередко обозначают с использованием суффикса «бластома»: нефробластома, нейробластома, ретиноблас- тома.
Н ейробластома. Это наиболее частая детская опухоль солидного строения, развивающаяся за пределами центральной нервной системы. Она поражает надпочечники, симпатические ганглии и составляет 14 % всех новообразований у детей (см. главу 26).
Частота нейробластомы, по различным оценкам, составляет от 6 до 8 случаев на 1 млн детей. Средний возраст заболевших 2 года; 85—90 % больных детей моложе 5 лет. У девочек нейро- бластома встречается несколько реже и имеет лучший прогноз, чем у мальчиков. Обнаружена наследственная предрасположенность к развитию нейробластомы (вероятно, в форме унаследованных мутаций в зародышевых клетках, обусловливающих индивидуальную восприимчивость к вторичным соматическим повреждениям). Частота развития этой опухоли повышена у близнецов и сибсов, а также при синдроме Видемана—Беквита и нейрофиброматозе [синдром Беквита—Видемана (J.В.Beckwith, H.-R.Wiedemann) — комплекс врожденных аномалий: гиперплазия почек, поджелудочной железы, яичек, надпочечников, большая масса тела и др.].
Нейробластома развивается из клеток неврального гребня. Наиболее частая (до 40 %) локализация — мозговое вещество надпочечника и параспинальные ганглии. Реже опухоль располагается в области таза, шеи, груди. У взрослых нейробластома локализуется в области головы, шеи, ног. Макроскопически определяется узел, размеры которого могут значительно варьировать. Некоторые нейробластомы четко отграничены от окружающих тканей и могут иметь тонкую капсулу, другие же обладают выраженным инфильтрирующим ростом и быстро прорастают окружающие ткани (почки, почечная вена, нижняя полая вена, аорта). На разрезе опухоль представлена мягкой сероватой тканью, напоминающей вещество мозга. В узлах крупных размеров нередки очаги некроза, кровоизлияния, обызвествления.
Гистологическая картина нейробластомы зависит от степени дифференцировки опухоли. В большинстве случаев опухоль состоит из мелких лимфоцитоподобных клеток с темными ядрами и скудной цитоплазмой, формирующих солидные пласты (рис.
А). Более дифференцированные опухолевые клетки имеют эозинофильные нейрофибриллярные отростки и располагаются в фибриллярной строме. В некоторых опухолях можно наблюдать формирование псевдорозеток в виде венчика клеток, окружающих эозинофильные скопления нейрофибрилл. При электронно-микроскопическом исследовании в опухолевых клетках определяются нейросекреторные гранулы и отростки с микротрубочками. Дальнейшая дифференцировка приводит к образованию элементов типа ганглиозных — крупных клеток с широким ободком эозинофильной цитоплазмы, большим пузырьковидным ядром и хорошо выраженными ядрышками (рис.
Б) (см. главу 26). В ткани опухоли увеличивается количество фибриллярной стромы. Опухоль с завершенной дифферен- цировкой представлена зрелыми ганглиозными клетками, окруженными пучками соединительной ткани, нервными волокнами и леммоцитами (шванновскими клетками). Такие опухоли называют ганглионейромами. Опухоль может содержать участки с разной дифференцировкой, поэтому диагноз ганглионейромы может быть поставлен лишь при анализе множества срезов из разных участков опухоли. Некоторые нейробластомы подверга-
Рис.
22.14. Нейробластома.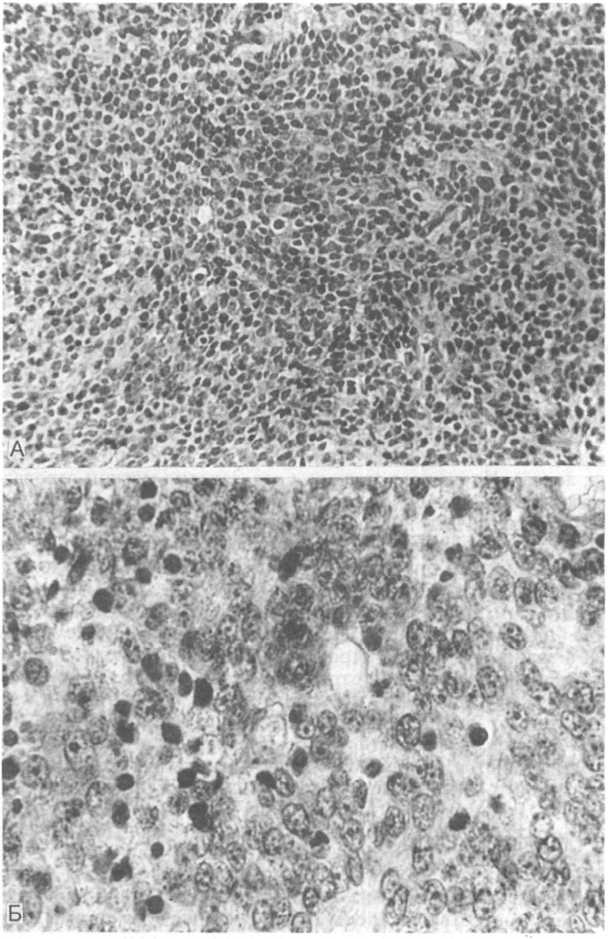
А — опухоль состоит из мономорфных клеток с темными ядрами и скудной цитоплазмой, между клетками небольшое количество нежного фибриллярного материала; Б — нейробластома с признаками дифференцировки, видны клетки с широким ободком цитоплазмы, пузырьковидными ядрами и хорошо выраженными ядрышками.
ются дифференцировке и дозревают до ганглионейром или спонтанно регрессируют. Регрессия чаще наблюдается при маленьких размерах опухоли. Метастазы отмечаются в 50 % случаев нейробластом у детей в возрасте до 1 года и в 70 % случаев у детей старшего возраста. Метастазы чаще всего обнаруживают в лимфатических узлах, костном мозге, костях, печени, подкожной клетчатке.
Международная классификация стадий роста нейробластомы следующая.
Стадия Характеристика опухоли
1 Опухоль, не выходящая за пределы органа, в котором она развилась. Метастазы отсутствуют. Новообразование удалено полностью
2а Односторонняя опухоль, распространяющаяся за пределы органа, в котором она развилась, но не пересекающая срединную линию. Удалена большая часть опухоли. Метастазы отсутствуют
2Ь Односторонняя опухоль любых размеров, удаленная полностью или неполностью, с метастазами в региональные лимфатические узлы
Неподдающаяся полной резекции опухоль, распространяющаяся через срединную линию независимо от вовлечения в процесс лимфатических узлов; односторонняя опухоль с метастазами в лимфатические узлы противоположной стороны тела; опухоль, расположенная по срединной линии с двусторонними метастазами в лимфатические узлы
Опухоль с метастазами в отдаленные лимфатические узлы и гематогенными метастазами
4S Опухоль, соответствующая стадиям 1 и 2 с гематогенными метастазами в кожу, печень, костный мозг у детей моложе 1 года
У маленьких детей новообразование, как правило, обнаруживают случайно при пальпации живота или при рентгенологическом обследовании по поводу каких-либо других заболеваний. У детей постарше в большинстве случаев опухоль диагностируют лишь при появлении отдаленных метастазов. Диагностика нейробластомы основывается на клинических и морфологических данных. Результаты биохимических и цитогенетических исследований могут помочь в диагностике, но они недостаточно специфичны. В крови у 90 % больных увеличена концентрация катехоламинов, соответственно повышена их экскреция с мочой. В клинической практике используют определение суточной экскреции ванилилминдальной и гомованилиновой кислот.
Прогноз при нейробластоме неоднозначен, он зависит от множества факторов. Существует ряд параметров и маркеров, которые могут помочь в определении прогноза опухоли. На прогноз во многом влияют такие показатели, как возраст ребенка и стадия роста опухоли. Наилучший прогноз имеют дети до 1 года независимо от стадии заболевания. Суммарно их выживаемость составляет 85—90 %, а у детей с нейробластомой 1—2 стадии (односторонняя опухоль без метастазов) она достигает 98 %. У детей старше 1 года прогноз значительно хуже. При наличии лимфогенных и гематогенных метастазов выживаемость не превышает 10 %.
Из прогностически важных маркеров следует назвать делецию короткого плеча хромосомы 1 дистальнее р32. Она приводит к потере супрессорного гена нейробластомы и существенно влияет на прогноз. Это наиболее типичная цитогенетическая аномалия при нейробластоме, однако она встречается и при других злокачественных опухолях. Описаны также случаи потери гете- розиготности длинного плеча хромосом 13 и 14. Значение такой генетической гетерогенности остается неясным. О неблагоприятном прогнозе свидетельствует также амплификация онкогенов N-myc и N-ras. Множественные копии N-myc (в некоторых случаях до 300) обнаруживаются при диссеминации опухоли. Для опухолей с доброкачественным течением амплификация N-myc нехарактерна. Однако в некоторых случаях с единственной копией наблюдается быстрое прогрессирующее течение. Таким образом, имеет значение не простое увеличение количества копий, а уровень экспрессии гена. Плоидность опухолевых клеток также влияет на прогноз. Гипердиплоидия сочетается с плохим прогнозом, анэуплоидия — с благоприятным. Показано также, что при высоком уровне экспрессии гена Trk выживаемость увеличивается. Дифференцировка нейробластов в более зрелые ганглиозные клетки частично происходит за счет действия фактора роста нервов, рецептор которого кодируется протоонкогеном Trk. Наконец, имеется несколько сывороточных маркеров, повышение уровня которых указывает на неблагоприятный прогноз. Это нейронспецифическая енолаза, ферритин, лактат- дегидрогеназа. Увеличенная концентрация ферритина в сыворотке наблюдается у 50 % больных со стадиями 3 и 4 болезни.
Ретинобластома. Это злокачественная опухоль глаза, встречается с частотой 1 на 20 000 новорожденных и составляет 2,5—4,5 % всех злокачественных опухолей у детей. Ретинобластома может быть одно- и двусторонней, однофокусной и многоочаговой. Она часто бывает врожденной. Ретинобластома, как и некоторые другие опухоли у детей, может подвергаться спонтанной регрессии. Описаны семейные и спорадические случаи ретино- бластомы. Ненаследственные ретинобластомы всегда односторонние и одноочаговые. На долю семейных (наследственных) случаев приходится около 50 %. Опухоль при этом обычно двусторонняя, часто многоочаговая. Больные с генетическими формами ретинобластомы имеют предрасположенность к развитию и других видов злокачественных новообразований. Большее число наблюдений ретинобластомы приходится на возраст до 4 лет.
Развитие ретинобластомы связано с мутацией гена Rb, расположенного в хромосоме 13 (13ql4). Этот ген кодирует ядерный белок, который блокирует выход клетки из Gl-фазы клеточного цикла, а также играет определенную роль в дифферен- цировке клеток. В генетически детерминированных случаях дети рождаются с одним нормальным и одним дефектным геном Rb, который унаследован от одного из родителей (семейные случаи) или стал результатом новой мутации в зародышевых клетках (генетические спорадические случаи). Мутация второго гена — соматическая, она происходит в сетчатке. При ненаследственных спорадических ретинобластомах обе мутации соматические. Таким образом, в клетке сетчатки, дающей начало ненаследственной ретинобластоме, должно возникнуть две соматические мутации. Поскольку уровень соматических мутаций низкий, у пациентов со спорадическими ретинобластомами имеется лишь единичный очаг опухоли. У пациентов с семейной ретинобластомой повышен также риск развития остеосарком и других опухолей мягких тканей. Инактивация гена Rb обнаружена и при мел ко клеточном раке легкого, раке мочевого пузыря, молочной и предстательной желез.
Ретинобластома развивается из клеток нейроэпителиального происхождения (см. главу 26). Опухоль располагается в задней камере глаза как одиночное или множественное сосочковое образование розовато-белого цвета с очагами некроза и петрифи- катами. Гистологически опухоль представлена мелкими округлыми клетками с гиперхромными ядрами и скудной цитоплазмой; иногда такие клетки образуют лентовидные структуры. Более дифференцированные формы ретинобластомы содержат кубические или невысокие призматические клетки, формирующие истинные розетки. Такие клетки содержат образования, напоминающие фоторецепторы. Вначале опухоль располагается в пределах сетчатки, но по мере роста она разрушает стекловидную пластинку, распространяясь на сосудистую оболочку и стекловидное тело и может заполнить всю полость глазного яблока, разрушить костные стенки глазницы. По ходу зрительного нерва опухоль способна прорастать в полость черепа. Ретинобластома метастазирует лимфогенно и гематогенно. Наиболее частая локализация лимфогенных метастазов — околоушные, подчелюстные, шейные лимфатические узлы, гематогенных метастазов — кости черепа, трубчатые кости конечностей и печень. Бывает спонтанная регрессия опухоли, сопровождающаяся обызвествлением и выраженной воспалительной реакцией.
Проявления ретинобластомы разнообразны. Первые клинические признаки болезни выражаются в беловатом свечении зрачка, расширении зрачка, ослаблении его реакции на свет и косоглазии. У больных снижается острота зрения. При прорастании сосудистой оболочки появляются боль в глазу, отек роговицы. Часто развивается воспалительный процесс (иридоцик- лит, увеит). Вследствие отека орбитальной клетчатки может возникнуть экзофтальм. При распространении ретинобластомы в полость черепа присоединяются мозговые симптомы. Успех лечения во многом зависит от возраста ребенка. У детей до 1 года смертность составляет 70 %, у детей постарше — около 20 %. Если ретинобластома ограничена пределами глаза, то имеются наибольшие шансы на выздоровление по сравнению с таковыми при других злокачественных опухолях. Ранняя диагностика значительно улучшает прогноз, при этом возможны органосохраняющие операции с сохранением зрения. При распространении ретинобластомы за пределы глазного яблока прогноз резко ухудшается. Больные с ретинобластомой умирают, как правило, от метастазов. Описаны случаи развития остеосаркомы глазницы после лучевой терапии ретинобластомы.
Опухоль Вильмса (M.Wilms; нефробластома). Это самая частая первичная опухоль почек детского возраста. Она составляет 7,8 случаев на 1 млн детей в возрасте 1 — 14 лет. Среди всех злокачественных новообразований у детей на долю нефробластомы приходится 6—7 %. Опухоль редко встречается у детей в возрасте до 6 мес и после 5 лет. Наибольшая заболеваемость наблюдается в возрасте 2—3 года.
Опухоль Вильмса может наблюдаться в виде спорадических, семейных и ассоциированных с синдромами форм. Большинство больных обладают нормальным кариотипом, однако иногда имеется связь с делецией 11р13, где расположен ассоциированный с опухолью ген WT-1. Ген WT-1 является геном-супрессором, его продукт — регулятор фактора транскрипции, связывающегося с ДНК. Белок WT-1 экспрессируется почками и гонадами плода (трансгенные мыши с отсутствием обоих копий ло- куса WT-1 имеют агенезию почек). У некоторых больных с опухолью Вильмса обнаружены мутация гена WT-2, расположенного в коротком плече хромосомы 11 дистальнее локуса гена WT-1, а также потеря гетерозиготности длинного плеча хромосомы 16.
Опухоль Вильмса часто ассоциируется с врожденными пороками развития. Наиболее постоянно это сочетание наблюдается при трех синдромах. WAGR-синдром (Wilms’ tumour, aniridia, genital anomalies, mental retardation) включает, как следует из названия, аниридию (отсутствие радужки или ее части), аномалии половых органов и умственную отсталость. Риск развития опухоли Вильмса у таких больных составляет 33 %. При WAGR- синдроме выявлена мутация аутосомно-доминантного гена с локализацией 11р13. Проксимальнее его, также в районе р13, располагается ген WT-1. Во многих случаях у пациентов с WAGR-синдромом имеется спорадическая делеция генетического материала в области 11р13, включающая оба эти локуса.
Синдром Дени—Драша (P.Denys, A.Drash) характеризуется дисгенезией гонад (мужским псевдогермафродитизмом) и нефропатией, приводящей к почечной недостаточности. У большинства таких больных развивается опухоль Вильмса; генетическая аномалия также локализована в 11р13, однако она представлена не делецией, а негативной доминантной мутацией гена WT-1.
Синдром Видемана—Беквита (J.В.Beckwith, H.R.Wiedemann) характеризуется увеличением размеров внутренних органов (висцеромегалией), гемигипертрофией, кистами мозгового слоя надпочечников, аномально большими клетками коры надпочечников и высоким риском развития опухоли Вильмса. У этих пациентов поврежден локус 11р15.5, расположенный дистальнее локуса гена WT-1 и названный геном WT-2. Функция гена WT-2 остается неясной. У больных со спорадическими случаями синдрома Видемана—Беквита выявлена потеря материнских аллелей в сочетании с отцовской дисомией в ло- кусе 11р15.5, что указывает на роль геномного импринтинга в генезе опухоли. У пациентов с синдромом Видемана—Беквита, кроме того, повышена частота развития гепатобластомы, рака коры надпочечников, рабдомиосарком, опухолей поджелудочной железы.
Макроскопически нефробластома, как правило, представлена большим одиночным узлом, четко отграниченным от ткани почки. Она имеет мягкую консистенцию, серовато-розовая, с очагами некроза, кровоизлияниями и кистами. Многоочаговое и двустороннее поражение встречаются в 10 % случаев. Микроскопически опухоль Вильмса представлена производными неф- рогенной ткани на разных стадиях дифференцировки. Для опухоли характерно сочетание трех компонентов: бластемного, эпителиального и стромального (рис. 22.15). Бластемный компонент представлен округлыми мелкими клетками с гиперхром- ными ядрами и узким ободком цитоплазмы. Эпителиальный компонент опухоли составляют различного вида трубочки, отражающие разные стадии дифференцировки нефрона, реже — клубочки. Мезенхимальный компонент представлен рыхлой незрелой соединительной тканью, в которой могут встречаться участки гладких и поперечнополосатых мышц, жировая ткань, хрящ и кости. Наличие в опухоли дифференцированных тканей не влияет на прогноз. Единственный гистологический признак, свидетельствующий о плохом прогнозе, — наличие в опухоли анаплазии. Применительно к опухоли Вильмса понятие анапла- зии означает появление клеток с увеличенными гиперхромны- ми, полиморфными ядрами и патологическими митозами. Me- тастазирует опухоль как лимфогенно, так и гематогенно. Лимфогенные метастазы направлены в региональные коллекторы ворот почки, парааортальные лимфатические узлы, далее в коллекторы ворот печени и брыжейки. Гематогенные метастазы обнаруживают преимущественно в легких. Метастазы в кости нехарактерны (в отличие от других опухолей почек). В метастазах, как правило, преобладают неэпителиальные компоненты опухоли.
Рис.
22.15. Опухоль
Вильмса. Островки недифференцированных
темных клеток бластемы расположены
среди примитивной мезенхимы. В очагах
бластемы наблюдается образование
трубочек.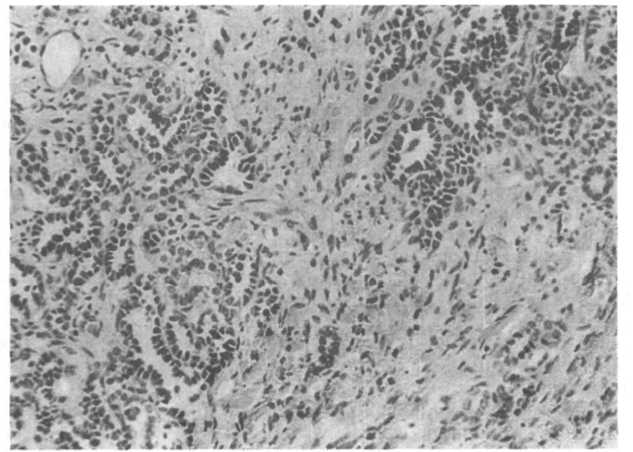
Во многих случаях опухоли Вильмса, а также различных врожденных и наследственных аномалий в почках находят очаги из примитивных, недифференцированных клеток, формирующих тубулярные метанефрогенные структуры. В отличие от опухоли Вильмса стромальные и эпителиальные структуры в них не встречаются. Фигуры митоза очень редки. Эти очаги называют нефробластоматозом. В почках, удаленных по поводу опухоли Вильмса, такие очаги обнаруживают в 20—44 % случаев. Их считают «предшественниками» опухоли Вильмса, однако известно, что в большинстве случаев из очагов персистируюшей бластемы опухоли не развиваются.
Как правило, опухоль Вильмса впервые проявляется как объемное образование в брюшной полости, случайно обнаруженное родителями или педиатром. Возможны боли в животе, обычно связанные с кровоизлиянием в опухоль, в 20 % случаев выявляется кровь в моче, чаще микрогематурия. Встречается непроходимость кишечника. У некоторых больных возникают симптомы, связанные с секрецией ренина опухолью: гипертензия, жажда и полиурия. Нередко уже при первичном выявлении опухоли обнаруживают метастазы в легкие. Применяют комбинированное лечение: хирургическое с пре- и постоперационной химио- и лучевой терапией
.Стадии опухоли Вильмса следующие.
Стадия Характеристика опухоли
Опухоль ограничена почкой и полностью удалена; капсула ин- тактна
Опухоль выходит за пределы почки, но полностью удалена; резекция проведена в пределах здоровых тканей
Остаточная опухоль в брюшной полости без гематогенных метастазов. Имплантационные метастазы в брюшину. Метастазы в лимфатические узлы за пределами парааортальной зоны. Элементы опухоли определяются в краях резецированного материала
Гематогенные метастазы или метастазы в лимфатические узлы
вне брюшной полости
Двусторонняя опухоль
При стадиях 1 и 2 излечение превышает 90 %, при стадии
4 — около 60 %. У детей со стадией 5 (двусторонняя опухоль) прогноз, против ожидания, тоже достаточно благоприятный.Глава 23 ЭНДОКРИННЫЕ ЗАБОЛЕВАНИЯ
Гомеостаз клеток регулируется нервной и эндокринной системами, интегрированными через гипоталамус. Последний модулирует активность гипофиза и широко распространенных в организме нейроэндокринных клеток, входящих в так называемую APUD-систему (см. главы 6, 16 и др.).
Гормоны, вырабатываемые эндокринными железами, взаимодействуют с органами-мишенями через рецепторы клеток, специфичных для определенных гормонов. Различают три основных типа рецепторов: поверхностные мембранные, цитоплазматические и внутриядерные.
Полипептидные (гипофизарные) и аминовые (например, ка- техоламиновые) гормоны взаимодействуют с рецепторами на поверхности клеток. Связывание гормона рецептором вызывает конформационные или биохимические изменения в рецепторе. Последние посредством протеинкиназы или сигнального передатчика протеина G активируют один или более вторичных передатчиков. К таким веществам относят циклический адено- зинмонофосфат, тирозинкиназу, мембранные фосфолипиды, внутриклеточный ионизированный кальций.
Стероидные гормоны проникают сквозь липидные мембраны клеток и взаимодействуют с рецепторами главным образом в цитоплазме, но иногда и в ядре. В любом случае комплекс гормон-рецептор после транслокации в ядро (в случае цитоплазматических рецепторов) связывает ДНК и активирует специфические гены, приводя к транскрипции и трансляции новых белков, которые обеспечивают действие гормонов.
Рецепторы тиреоидных гормонов локализованы в основном в ядре и непосредственно связаны с ДНК, но некоторые из них могут находиться и в цитоплазме. Вначале гормоны связывают участки на поверхности клеток, затем комплексы белок—гормон проникают в ядра или цитоплазму. Комплекс гормон—рецептор активирует транскрипцию специфических генов таким же способом, как и в случае стероидных гормонов.
Функция эндокринных желез контролируется путем взаимодействия стимулирующих и тормозящих гормонов, образующихся в гипоталамусе (гипофизе), или с помощью механизма обратной связи (периферические эндокринные железы, находящиеся под контролем гипофиза). Например, когда уровень кортизола недостаточен для поддержания гомеостаза, тормоз
ное действие кортизола на секрецию кортикотропин-рилизинг- гормона (КРГ) и адренокортико- тропного гормона (АКТГ) увеличивается, приводя к усилению выброса АКТГ и синтеза кортизола (схема 23.1). Когда количество кортизола нормализуется, синтез КРГ и АКТГ подавляется. Этот гормональный механизм обратной связи может обеспечиваться и иными факторами, чем гормоны, например биосинтез кортизола регулируется концентрацией катионов в сыворотке крови, осмолярностью сыворотки и объемом внеклеточной жидкости.
Схема
23.1.
Механизм
обратной связи: взаимодействие между
гипоталамусом, гипофизом и надпочечником
Гипоталамус

Гипофиз состоит из двух долей, различающихся по эмбриогенезу, морфологии и функции.
Передняя доля гипофиза (аденогипофиз) построена из секреторных эпителиальных клеток. По особенностям окрашивания цитоплазмы клеток гематоксилином и эозином их делят на три типа. ацидофилы, базофилы (хромофилы) и хромофобы. Иммуногистохимически, используя моноклональные антитела, можно выявить 5 типов клеток, продуцирующих тропные гормоны: 1) соматотропы (продуцируют гормон роста); 2) маммотропы (продуцируют пролактин) — ацидофилы; 3) меланокортикотро- пы (продуцируют проопиомеланокортин — предшественник АКТГ, меланоцитстимулирующего гормона, липотропина, (3-эн- дорфина); 4) тиреотропы (продуцируют тиреоидстимулирующий гормон); 5) гонадотропы (продуцируют фолликулостимулирующий и лютеинизирующий гормоны) — базофилы. Хромофобы, клетки со слабо окрашивающейся цитоплазмой, содержат небольшое количество секреторных гранул, поэтому иммуногистохимичес- ким методом определить в них гормоны трудно. Функциональная активность секреторных клеток регулируется гипоталамусом.
Тиреоидстимулирующий, фолликулостимулирующий и лютеинизирующий гормоны — гликопротеины, молекула которых состоит из а-субъединиц, одинаковых у всех трех гормонов, и из различных р-субъединиц, специфичных для каждого гормона. Иногда эти субъединицы обнаруживают в крови. При электронно-микроскопическом исследовании все секреторные клет
ки содержат гранулы диаметром 50—1000 нм, окруженные мембраной и содержащие тропный гормон.
Задняя доля гипофиза (нейрогипофиз) состоит из переплетенных немиелинизированных нервных волокон, содержащих секреторные гранулы, которые заполнены гормонами вазопресси- ном, или антидиуретическим гормоном (АДГ), и окситоцином. Эти гормоны синтезируются в супраоптическом и паравентри- кулярных ядрах гипоталамуса и транспортируются по нервным волокнам в заднюю долю гипофиза.
Заболевания гипофиза делят на те, при которых в процесс первично вовлечена передняя доля, и те, при которых поражена задняя доля. Для заболеваний с преимущественным поражением аденогипофиза характерны: повышенная или сниженная продукция тропных гормонов. Повышенный синтез тропных гормонов расценивают как гиперпитуитаризм и чаще всего связывают с развитием функционирующей опухоли передней доли гипофиза, реже он бывает связан с торможением механизма обратной связи на уровне гипоталамуса. 75 % случаев гипопитуи- таризма связано с разрушением передней доли гипофиза нефункционирующей опухолью. Гипопитуитаризм редко бывает гипоталамического происхождения.
Для поражений аденогипофиза характерны также местные проявления:
увеличение турецкого седла, связанное с внешними воздействиями (например, опухолью гипофиза), обнаруженное при рентгенологическом исследовании, компьютерной или магнитно-резонансной томографии;
нарушение полей зрения, вызванное давлением опухоли гипофиза на зрительный перекрест или зрительные нервы;
признаки повышенного внутричерепного давления в виде головной боли, тошноты и рвоты, связанные с развитием крупной опухоли гипофиза.
Все заболевания передней доли гипофиза делят на две большие группы: гиперпитуитаризм — опухоли передней доли; заболевания, связанные с гипопитуитаризмом.
Гиперпитуитаризм. Аденомы. Гиперфункция аденогипофиза чаще всего обусловлена развитием аденом. Функционально активные (продуцирующие гормоны) карциномы и заболевания гипоталамуса, вызывающие стимуляцию гипофиза, встречаются редко.
Некоторые аденомы продуцируют более одного гормона. Иногда представлены сразу две клеточные популяции — сома- тотропы и маммотропы. Встречаются аденомы, состоящие из предшественников клеток гипофиза. Они выделяют предшественники гормонов. В большинстве случаев клинические проявления опухоли обусловлены лишь одним гормоном. Встречают - ся нефункционирующие аденомы, которые, обладая экспансивным ростом, разрушают переднюю долю гипофиза и приводят к гипопитуитаризму.
Аденомы гипофиза делят на микроаденомы (менее 10 мм в диаметре) и макроаденомы (более 10 мм в диаметре). Микроаденомы широко распространены и встречаются в 40 % патологоанатомических исследований. В большинстве случаев эти опухоли являются случайными находками. Хотя микроаденомы в аденогипофизе чаще бывают единичными, встречаются и множественные аденомы. Микроаденомы всегда локализованы в самом гипофизе. Макроаденомы могут полностью занимать турецкое седло и ограниченную часть передней доли гипофиза, иногда при выраженном экспансивном росте они вытесняют весь аденогипофиз и даже заднюю долю, разрушают окружающие кости и сдавливают соседние структуры. Аденомы, ограниченные турецким седлом, плохо инкапсулированы. Крупные, быстро растущие опухоли могут проникать сквозь капсулу в соседние структуры, такие как зрительный перекрест, соседние черепные нервы, основание мозга, кавернозные синусы и клиновидную кость.
Микроскопически все аденомы имеют похожее строение. Более или менее одинаковые полигональные клетки образуют структуры в виде листов, тяжей или гнезд и содержат тонкую васкуляризованную строму. Иногда встречаются псевдожеле- зистые или папиллярные структуры. Обнаруживают мелкие или крупные фокусы ишемического некроза, а также пеаммомные тельца и кровоизлияния. При электронно-микроскопическом исследовании в цитоплазме опухолевых клеток выявляются многочисленные секреторные гранулы.
Соматотропные аденомы являются основной причиной акромегалии и гигантизма. При избыточном образовании гормона роста у взрослых развивается акромегалия. Она характеризуется увеличением выступающих частей тела: кистей и стоп, челюсти, языка, а также мягких тканей. Эти изменения развиваются в течение десятилетий, прежде чем будут распознаны, а аденома достигнет существенных размеров. Если соматотропные аденомы появляются у детей прежде, чем закроются эпифизы, возникает гигантизм. В настоящее время гигантизм наблюдается чрезвычайно редко. Около 50 % соматотропных аденом состоит из сильно гранулированных зрелых клеток, которые интенсивно окрашиваются на гормон роста при иммуногистохимическом исследовании. Другие 50 % аденом чаще всего состоят из полиморфных ацидофилов, в которых наблюдается слабая реакция на гормон роста. В этих случаях признаки акромегалии могут быть слабовыражены, преобладают местные проявления опухоли гипофиза.
Около 30 % аденом, продуцирующих гормон роста, бигормо- нальные и одновременно вырабатывают пролактин. В болыиин- стве этих аденом обнаруживают соматотропы и маммотропы. Реже эти опухоли бывают мономорфными и состоят из предшественников двух разных типов клеток.
Пролактиномы. Аденомы, секретирующие пролак- тин, — наиболее распространенная опухоль гипофиза. Гипер- пролактинемия может вызывать гипогонадизм как у мужчин, так и у женщин, галакторею — у женщин. Эта опухоль наблюдается у 25 % больных аменореей. У мужчин могут развиться импотенция и бесплодие.
Около 60 % пролактинсекретирующих опухолей являются микроаденомами, состоящими из сильно гранулированных ацидофильных клеток. Остальные опухоли — макроаденомы. Действительно, большинство гипофизарных опухолей, обнаруженных при патологоанатомическом исследовании, состоят из клеток, которые синтезируют пролактин. Иногда пролактиномы при обычной окраске гематоксилином и эозином могут быть хромофобными или незначительно ацидофильными. Для идентификации тропного гормона, содержащегося в гранулах, необходимо использовать иммуногистохимические методы.
Гиперпролактинемия в редких случаях может формироваться за счет гиперплазии маммотропов гипофиза без развития аденом, например при поражении гипоталамуса, приеме веществ, нарушающих допаминергическую передачу (метилдопа, резерпин), и лечении эстрогенами.
Кортикотропные опухоли. Большинство корти- котропных опухолей представлено базофилъными микроаденомами. Усиление продукции АКТГ ведет к гиперсекреции кортизола надпочечниками и развитию болезни Кушинга. Более редким вариантом является хромофобная кортикотропная аденома; при ней симптомы избыточного образования кортизола выражены слабее. Эти «молчащие» опухоли чаще бывают крупными и обусловливают развитие местных изменений. В классических базо- фильных кортикотропных аденомах при проведении иммуно- гистохимических реакций наблюдают выраженную реакцию на АКТГ.
Достаточно редко повышенное образование АКТГ связано с гиперплазией кортикотропов или множественными микроаденомами.
Другие функционально активные адено- м ы. Гонадотропные аденомы встречаются примерно в 6 % случаев опухолей гипофиза. У мужчин с такой опухолью в сыворотке крови увеличено содержание фолликулостимулирующего гормона и незначительно повышен уровень лютеинизирующего гормона. Клинически заболевание проявляется гипогонадизмом. В большинстве случаев обнаруживают гиперпродукцию р-субъ- единиц фолликулостимулирующего гормона. У женщин часто не бывает признаков увеличения синтеза гонадотропных гормонов, хотя в сыворотке крови определяется лютеинизирующий гормон. Как у мужчин, так и у женщин опухоли чаще всего крупные и сопровождаются местными проявлениями. Тирео- тропные аденомы встречаются редко.
Карциномы. Аденокарциномы аденогипофиза встречаются редко, большинство из них функционально неактивно. Они бывают хорошо дифференцированными и напоминают атипичные аденомы или плохо дифференцированными с разной степенью полиморфизма. Карциномы аденогипофиза, как правило, распознают после появления метастазов (обычно в лимфатические узлы, кости, печень).
Заболевания, связанные с гипопитуитаризмом. Гипофункция аденогипофиза может быть обусловлена поражением гипоталамуса или самой передней доли гипофиза. Повреждение гипоталамуса встречается очень редко и вызвано развитием супрасел- лярной краниофарингиомы, глиомы или опухоли из зародышевых клеток. Такие опухоли обусловливают различные клинические синдромы, включая несахарный диабет, ускоренный рост, задержку роста и созревания.
Около 90 % случаев гипопитуитаризма связано с деструктивными процессами, затрагивающими аденогипофиз. К трем наиболее распространенным повреждениям относят несекретирующие аденомы, некроз гипофиза (синдром Шихана; H.L.Sheehen) и синдром пустого турецкого седла. Другие случаи гипофизарной недостаточности обусловлены метастатическими опухолями, кровоизлиянием в гипофиз, нарушением кровообращения в гипофизе в результате системного артериита или тромбоза кавернозных венозных синусов, воспалительной деструкцией передней доли при саркоидозе или инфекциях, хирургическим либо радиационным повреждением гипофиза. Гипофункция гипофиза проявляется после разрушения по крайней мере 75 % его передней доли.
Клинические проявления деструктивных повреждений аденогипофиза исключительно разнообразны. В препубертатном возрасте наблюдается симметричная задержка роста, так называемая гипофизарная карликовость, и полового развития. У взрослых отсутствие гормона роста per se можно распознать лишь путем определения гормонального профиля с помощью радио- иммунных методов. Гипогонадизм у женщин сопровождается аменореей, исчезновением волос на лобке и в подмышечных впадинах, стерильностью и атрофией яичников и наружных половых органов. У мужчин гипогонадизм проявляется атрофией яичек, стерильностью и отсутствием волос на лобке и в подмышечных впадинах. Все эти изменения связаны со снижением функции половых желез. Встречается гипотиреоидизм, обусловленный отсутствием тиреоидстимулирующего гормона, а также гипоадренализм, сопровождающийся дефицитом АКТГ, атрофией щитовидной железы и надпочечников. Таким образом, недостаточность щитовидной железы, надпочечников или половых желез, связанную с отсутствием тропных гормонов гипофиза, необходимо дифференцировать от первичных заболеваний этих органов. Деструктивный процесс может затрагивать и заднюю долю гипофиза. Пангипопитуитаризм встречается редко и сопровождается резким уменьшением массы тела (кахексия Симмондса; M.Simmonds).
Несекретирующие хромофобные аденомы гипофиза. Около 25—30 % диагностированных опухолей гипофиза клинически функционально неактивны. Больные обращают внимание на так называемые местные проявления: нарушение полей зрения, головные боли или гипофункция одного либо нескольких эндокринных органов-мишеней, находящихся под контролем гипофиза (например, гипотиреоидизм или гипогонадизм). Обычно эти аденомы в момент обнаружения уже крупные, так как в течение многих лет клинически они не проявлялись. При общем обследовании их не удается дифференцировать от секретирующих аденом.
Гистологически большинство таких аденом можно классифицировать как «нулевые» или онкоцитомы. Первые состоят из клеток, в которых секреторные гранулы совершенно отсутствуют или встречаются в небольших количествах. При иммуногистохими- ческом исследовании во многих клетках обнаруживают фолликулостимулирующий гормон, свободные а- и р-субъединицы или (реже) лютеинизирующий гормон. Онкоцитомы состоят из клеток с обильной эозинофильной зернистой цитоплазмой, богатой митохондриями. Полагают, что онкоцитарные клетки образуются из слабогранулированных или нулевых клеток.
Синдром Шихана. Синдром известен как послеродовый некроз гипофиза. Развивается в результате остро возникшей ишемии передней доли гипофиза при маточном кровотечении или шоке. Во время беременности гипофиз увеличивается почти в 2 раза, сдавливая питающие его кровеносные сосуды. Внезапное системное снижение артериального давления приводит к спазму сосудов, и таким образом развивается ишемический некроз большей части или всей передней доли гипофиза. При этом задняя доля сохраняется, так как она менее чувствительна к аноксии.
В развитии синдрома Шихана могут участвовать и другие патогенетические механизмы, такие как диссеминированное внут- рисосудистое свертывание или (редко) серповидно-клеточная анемия, тромбоз кавернозных синусов, височный артериит или травматические повреждения сосудов. Опасность развития синдрома увеличивается у больных, длительное время страдающих сахарным диабетом.
Аденогипофиз при патологоанатомическом исследовании мягкий, бледный, ишемизированный или с кровоизлияниями. Ишемизированная зона со временем рассасывается и замещается фиброзной тканью.Синдром пустого турецкого седла. Это редкое заболевание, чаще связанное с попаданием в место, где расположен гипофиз, мягкой мозговой оболочки через дефект в диафрагме турецкого седла или слишком большое отверстие, через которое в норме проходит стебель гипофиза. Под давлением цереброспинальной жидкости развивается атрофия гипофиза, поэтому турецкое седло выглядит пустым. Другими причинами развития синдрома пустого турецкого седла являются синдром Шихана, инфаркт аденомы гипофиза с последующим рубцеванием, хирургическое удаление железы или ее облучение.
Супраселлярные опухоли гипоталамуса. Они могут вызывать гипофункцию или гиперфункцию передней доли гипофиза, несахарный диабет или комбинацию этих проявлений заболевания. Чаще встречаются глиомы и краниофа- рингиомы.
Краниофарингиома развивается из остатков кармана Ратке (M.H.Rathke; выпячивание эпителия задней стенки ротовой полости зародыша на границе с глоткой, которое является зачатком аденогипофиза), иногда располагается в турецком седле, но в основном супраселлярно и встречается у детей и молодых людей. Обычно является доброкачественной опухолью. Кранио- фарингиомы имеют диаметр 3—4 см, могут быть инкапсулированы и иметь солидное строение, однако чаще всего они кистозного вида. Более 75 % этих опухолей содержат выявляемые рентгенологически кальцификаты. Опухоль нередко давит на зрительный перекрест или зрительные нервы, реже на дно III желудочка и основание мозга.
Строение опухоли очень вариабельное. Она напоминает эмалевый орган зубов, поэтому эти опухоли известны также как адамантиномы или амелобластомы. Гнезда из тяжей многослойного плоского или цилиндрического эпителия разделены слаборазвитой фиброзной стромой. Часто гнезда плоских клеток вторгаются в периферические слои цилиндрических клеток. В кистозных вариантах опухоли выстилающий многослойный плоский или цилиндрический эпителий может быть гладким или образует сосочковые выросты. Кальцификация и формирование костной ткани наблюдаются в некротизированных центрах солидной опухоли так же, как и в клетках. Иногда в эпителиальных клетках встречаются анапластические изменения, однако рак развивается редко.
Синдромы задней доли гипофиза. Заболевания, связанные с дисфункцией задней доли гипофиза, встречаются редко. Последствия дисфункции этой доли проявляются в форме дефицита антидиуретического гормона, что приводит к развитию несахарного диабета, который характеризуется полиурией, чрезмерной жаждой и полидипсией. Причинами этого синдрома являются:
неопластические или воспалительные изменения гипотала- мо-гипофизарной области (например, супраселлярные опухоли,
метастазы рака, абсцессы, менингит, туберкулез и саркоидоз); 2) хирургическое или радиационное повреждение гипоталамо- гипофизарной системы (например, хирургическая или радиационная гипофизэктомия); 3) тяжелые повреждения головы. Встречаются также идиопатические случаи.