

Изменения центральной нервной системы при старении, дегенеративных процессах и деменции (слабоумии)
Как правило, в норме до 65-летнего возраста у взрослых масса мозга составляет 1200—1600 г. После 65 лет головной мозг нередко уменьшается из-за происходящих в нем атрофических процессов, причем полушария несколько отступают от костей черепа. При патологоанатомическом исследовании можно видеть, что оболочки мозга (особенно над теменной зоной) утолщенные с молочным отливом. В самом головном мозге обнаруживают более плотные и несколько суженные извилины, а также более широкие борозды, особенно в лобных и височных долях. На разрезе определяются некоторое истончение коры больших полушарий, уменьшение количества белого вещества и расширение системы желудочков. Под микроскопом при тщательных количественных исследованиях можно выявить небольшую утрату нейронов. В сером веществе возникают немногочисленные старческие (сенильные) бляшки, которые представляют собой бесструктурные скопления межклеточного вещества, окруженные сплетениями утолщенных аксонов и ядрами макро- и микроглии. Эти бляшки обнаруживаются преимущественно в лобных долях коры большого мозга, а также в гиппокампе и реже в подкорковых ядрах. Иногда видны признаки зернисто-вакуольной дегенерации, нейрофибриллярные пучки (в цитоплазме нейронов) или дегенерация нервных волокон. Мозжечок, как правило, остается интактным.
Деменции. Они представляют собой общие расстройства высшей нервной деятельности, которые развиваются у лиц, иногда не имеющих какой-либо соматической патологии. Деменции могут возникать при более или менее обширных явлениях деструкции либо дезорганизации в коре мозга, белом веществе или подкорковых ядрах. Эти поражения, представленные либо одним вариантом, либо сочетанием разных вариантов, вызываются тоже либо одним, либо несколькими патологическими процессами.
Перечислим причины деменции (слабоумия).
Первичные деменции
Болезни Альцгеймера, Пика, Хантингтона (Гентингтона), . Паркинсона
Вторичные деменции
Сосудистая патология: множественные инфаркты головного мозга, системная красная волчанка
Черепно-мозговые травмы: посттравматическая энцефалопатия, субдуральная гематома
Инфекции: нейросифилис — общий паралич у душевнобольных, постэнцефалические синдромы, болезнь Крейтцфельда—Якоба, СПИД
Гидроцефалия при нормальном внутричерепном давлении
По перечисленным причинам различают группы первичных деменций, при которых основные изменения происходят в коре (лишь иногда отмечается дегенерация в экстрапирамидном и спинальном трактах), и вторичных деменций, связанных с какими-либо тяжелыми первичными поражениями мозга — инфарктами, опухолями, прогрессивным параличом (тяжелое психическое заболевание, возникающее, как правило, при паретичес- кой форме нейросифилиса), тяжелыми интоксикациями и метаболическими нарушениями, тяжелыми инфекциями и гидроцефалией.
Первичные деменции. Болезнь Альцгеймера (болезнь Альцхаймера; A.Alzheimer) — наиболее распространенное дегенеративное поражение центральной нервной системы. Женщины болеют чаще мужчин. Этиология и патогенез этого заболевания нуждаются в более подробном изучении.
При болезни Альцгеймера снижается холинергическая иннервация в зонах новой коры и гиппокампа, а в холинергичес- ких ядрах переднего мозга исчезают нейроны. В качестве возможных этиологических агентов рассматривают белок р-амилоид (Abeta) и его предшественник АРР (белок — предшественник амилоида, см. главу 5). В молекуле предшественника есть большой внеклеточный домен, единственный мембраносвязанный район и короткий внутриклеточный домен. Зона Ар относится к району, простирающемуся от внеклеточной «порции» молекулы к трансмембранному домену. У некоторых кровных родственников, страдающих семейной формой болезни Альцгеймера, заболевание, возможно, связано с точковой мутацией в кодирующем районе гена АРР. Другая точковая мутация, обнаруженная у представителей нескольких голландских родословных, обусловливает ускоренное отложение амилоида в стенках сосудов мозга. При этом появляется риск кровотечений, что нашло отражение в названии указанной формы болезни — «наследственное мозговое кровоизлияние с амилоидозом». Определенные изоформы амилоида содержат домен ингибитора протеазы, и роль этой молекулы в регуляции каскадного формирования белкового сгустка рассматривают как возможную ведущую функцию АРР. Выходе нормальной «переработки» АРР в середине последовательности молекулы Ар расщепляется пептидная связь, в результате чего предотвращается образование нерастворимых агрегатов Ар. Таким образом, предполагают, что аномальная переработка молекулы предшественника, приводящая к накоплению возрастающих количеств пептида, лежит в основе отложений амилоидных масс. Кроме того, в тех клетках, в которых экспрессированы мутации, приводящие к семейной форме болезни Альцгеймера, обнаружено повышенное воспроизведение амилоидсоздающих Ар. У членов некоторых семей локус семейной формы болезни Альцгеймера располагается отдельно от гена АРР на хромосоме 21 или на других хромосомах.
Как правило, клинические признаки начинают проявляться с незаметного снижения высшей нервной (умственной) деятельности в виде изменений в настроении и поведении. Позднее появляются более тяжелые изменения: прогрессирующая утрата ориентации и памяти, афазия (нарушения речи, утрата способности понимать чужую речь и выражать свои мысли). Все это свидетельствует о тяжелых корковых поражениях доминантного полушария головного мозга. Через 5—10 лет больной полностью теряет трудоспособность, становится безмолвным и лишенным подвижности. Болезнь редко проявляется до 50-летнего возраста. Рост заболеваемости у лиц старше 50 лет вызывает беспокойство и повышает исследовательскую активность в тех странах, где увеличивается число старых людей. Так, в США, Англии и некоторых других англоязычных странах заболеваемость болезнью Альцгеймера в последнее десятилетие XX в. изменялась следующим образом: 3 % — в возрасте 65—74 года, 19 % — 75—84 года и 47 % — 85 лет и более. В большинстве случаев отмечают спорадические случаи, но у 5—10 % больных выявляют семейные формы. У лиц с трисомией 21, живущих дольше 45 лет, обнаружены те же морфологические изменения в центральной нервной системе, которые характерны для болезни Альцгеймера.
При патологоанатомическом исследовании в процессе осмотра головного мозга обнаруживают явления атрофии коры, выраженные в разной степени и сопровождающиеся расширением борозд. Эти явления больше выражены в лобных, височных и теменных долях. При значительной атрофии отмечается расширение желудочков, имеющее явно вторичный характер по отношению к утрате вещества мозга. Важнейшими патогистологическими признаками болезни Альцгеймера являются нейрофиб- риллярные пучки, старческие (сенильные) или нейритные бляшки и амилоидная ангиопатия. У тех пожилых пациентов, которые не имеют признаков деменции, указанные нарушения выражены в меньшей степени. Диагностика болезни Альцгеймера строится на клинико-патологических сопоставлениях неврологического состояния больного и частоты морфологических находок старческих бляшек и нейроФибриллярных пучков.
Нейрофибриллярные пучки представляют собой скопления филаментов в цитоплазме нейронов. Эти пучки располагаются возле ядер, иногда окружая и даже смещая их к плазмолемме. Они часто имеют вытянутую (в виде язычка пламени) или(реже) шаровидную форму. При окраске гистологических срезов гематоксилином и эозином нейрофибриллярные пучки выглядят как базофильные фибриллярные структуры. Они выявляются гораздо лучше после импрегнации срезов раствором нитрата, а затем аммиачного серебра по Билыновскому (M.Bielschowsky) с последующим восстановлением в формалине. Как правило, эти пучки определяются в корковых нейронах, особенно в энтори- нальной области (участке старой коры в парагиппокампальной извилине), пирамидных клетках гиппокампа, нейронах миндалин мозжечка, переднего мозга и ядрах шва (расположенных по средней линии продолговатого мозга). В электронном микроскопе пучки имеют вид спаренных извитых филаментов, перемежающихся с прямыми филаментами. Главным компонентом спаренных извитых филаментов являются аномально гиперфос- форилированные формы белка «тау» — аксонного протеина, который увеличивает агрегат микротрубочек. Другие антигены, обнаруженные в спаренных извитых филаментах, включают МАР2 (белок, связанный с микротрубочками), убиквитин и Ар. Спаренные извитые филаменты выявлены также в дистрофич- ных нейритах, формирующих внешние слои нейритных бляшек, а также в аксонах, проходящих через серое вещество в виде ней- ропильных нитей (нейропиль — переплетение нервных волокон). Несмотря на то что нейрофибриллярные пучки характерны для болезни Альцгеймера, они встречаются не только при этом заболевании, но и при прогрессирующем супрануклеарном параличе, постэнцефалитной болезни Паркинсона (см. ниже) и комплексе, состоящем из бокового амиотрофического склероза, паркинсонизма и деменции. По-видимому, указанные пучки представляют собой результат нескольких различных патологических процессов на клеточном уровне. Во всяком случае при болезни Альцгеймера они отражают аномальную организацию элементов цитоскелета нейронов.
Нейритные бляшки — очаговые сферические скопления утолщенных извилистых и воспринимающих соли серебра аксонов (дистрофических нейритов), окружающих центрально расположенный амилоидный стержень (рис. 26.9, А—Г). Вокруг этого стержня нередко определяется светлая зона. Нейритные бляшки встречаются при болезни Альцгеймера, их диаметр варьирует от 20 до 200 мкм. По периферии бляшек можно наблюдать клетки микроглии и астроциты. Бляшки обнаруживают в гиппокампе, миндалинах мозжечка, реже в новой коре, хотя двигательная и чувствительная зоны коры остаются относительно интактными. Небольшое число бляшек иногда имеется у пожилых лиц без деменции, поэтому при распознавании болезни Альцгеймера следует учитывать прежде всего количество бляшек. Дистрофические нейриты содержат спаренные извитые филаменты, а также синаптические везикулы и аномальные митохондрии. Амилоидный стержень, который окрашивается конго красным, солями серебра (по Билыновскому) и тиофлавином S, содержит разнообразные белки. Доминирующим компонентом стержня в бляшке является А(3, который представляет собой пептид, состоящий примерно из 40—43 аминокислотных остатков и образующийся из АРР (см. выше). Нередко отложения амилоидного пептида не сопровождаются реакцией нейритов. Такие поражения, названные диффузными бляшками, выявляются в поверхностных частях коры большого мозга, базальных ганглиях и коре мозжечка (рис. 26.9, Д, Е). Они формируются в основном возле мелких сосудов или пучков нейронов.
Амилоидная ангиопатия почти всегда сопровождает болезнь Альцгеймера, причем иногда она встречается при отсутствии других признаков болезни Альцгеймера. Амилоидные массы, находящиеся в сосудистых стенках, имеют тот же химический состав, что и амилоидные стержни в бляшках, описанных выше [по MacSween R.N.M., Whaley К., 1994]. Кроме того, отложения амилоида могут быть обнаружены в тканях, не относящихся к нервной системе.
Под гранулярно-вакуольной дегенерацией подразумевают формирование в цитоплазме нейронов мелких (менее 5 мкм в диаметре) светлых вакуолей, каждая из которых содержит аргиро- фильную гранулу (аргирофильность — это способность элементов ткани восстанавливать серебро из его солей в присутствии восстановителей). При болезни Альцгеймера и некоторых других заболеваниях центральной нервной системы выявляются также тельца Хирано (A.Hirano). Это вытянутые стекловидные эозинофильные тельца, состоящие из паракристаллических масс филаментов, похожих на бусы, в которых главным компонентом является актин. И дегрануляция, и тельца Хирано обна-
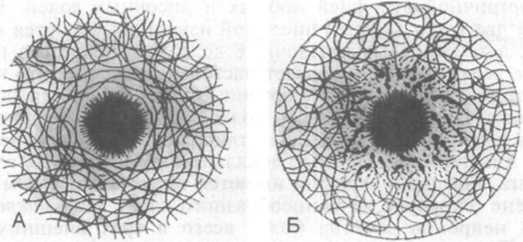
Рис.
26.9. Болезнь Альцгеймера.
А,
Б, В и Г (верхний ряд) — схематическое
изображение морфогенеза нейрит- ных
(старческ!
В
Ф. Шефера).
ных
(старческих) бляшек; Д и Е (нижний ряд)
— диффузные бляшки (негативы 3 Ф.Шефера).
руживают чаще всего в пирамидных клетках гиппокампа. Их значение неизвестно.
Болезнь Пика (A. Pick) встречается гораздо реже болезни Альцгеймера. При болезни Пика тоже возникает тяжелая деменция. Головной мозг характеризуется выраженной и нередко асимметричной атрофией лобных и височных долей. Причем задние две трети верхней височной извилины остаются интакт- ными, а теменные и затылочные доли вовлекаются в процесс крайне редко. Атрофия бывает настолько тяжелой, что извилины истончаются и приобретают вид тонкой вафли или даже острия ножа. При этом выражен долевой характер атрофии — ло- барная атрофия, позволяющая отличать это заболевание от болезни Альцгеймера. Помимо локальной атрофии коры головного мозга, признаки атрофии имеются также в хвостатом ядре и путамене (скорлупе чечевицеобразного ядра). Под микроскопом утрата нейронов заметна более всего в трех внешних слоях коры. По степени тяжести она может напоминать поверхностный (псевдо)ламинарный некроз (см. начало этой главы). Некоторые из выживающих нейронов, так называемые клетки Пика, имеют характерное набухание или содержат в своем перикарио- не тельца Пика. Эти тельца являются округлыми филаментны- ми включениями, обладающими слабой эозинофилией, но выраженной аргирофилией. Под электронным микроскопом видно, что они построены из нейрофиламентов, везикулярной эндо- плазматической сети и спаренных спиральных филаментов, сходных по иммуногистохимическим свойствам с аналогами, обнаруживаемыми при болезни Альцгеймера. Однако тельца Пика не являются маркерами болезни после гибели нейронов.
Болезнь Хантингтона (хорея Гентингтона; G.S.Huntington) наследуется по аутосомно-доминантному типу. При этом заболевании прогрессирующая деменция сопровождается непроизвольными хорееподобными движениями (хорея — это общее название гиперкинезов, т.е. автоматических насильственных движений вследствие непроизвольных сокращений скелетных мышц). Болезнь Хантингтона, как правило, начинается в 3-м или 4-м десятилетии жизни. Заболеваемость не превышает 4—7 случаев на 100 ООО человек. Патогенез остается неизвестным, но в последние годы у больных были выявлены аберрации в коротком плече хромосомы 4. При патологоанатомическом исследовании головной мозг обычно выглядит маленьким, наиболее примечательным признаком служит селективная атрофия хвостатого ядра. При болезни Хантингтона поражается несколько ней- ротрансмиттерных систем. Наиболее важными являются снижение количества у-аминомасляной кислоты (ГАМК), а также связанных с ней ферментов (например, декарбоксилазы глутаминовой кислоты).
Вторичные деменции. Слабоумие при множественных инфарктах мозга (мультиинфарктная деменция). Большинство таких больных — люди пожилого возраста, страдающие гипертонией. У них деменция отличается постепенной эволюцией. Нередко сильно развитые атеросклеротические бляшки, стенозирующие главные артерии головного мозга, приводят к повторным инфарктам. Какого-либо соответствия локализации инфарктов и характера деменции не выявлено. По-видимому, слабоумие развивается тогда, когда деструкции подвергается значительный объем вещества мозга, возможно, равный объему 100 мл ликвора. Обнаруживают расширенные желудочки. Такое расширение может быть симметричным, если множественные и мелкие инфаркты локализуются в каждом полушарии, и асимметричным в случае преимущественного поражения одного полушария.
Деменция у лиц с гидроцефалией при нормальном внутричерепном давлении. Она проявляется в виде расторможенного лобно-долевого типа с неотложными позывами на мочеиспускание и выраженной дискоординированной походкой.
Деменция при медленных вирусных инфекциях. Она сравнительно редко бывает связана с болезнью Крейтцфельда—Якоба — одной из ее форм, сопровождающейся спонгиоформной энцефалопатией.