

Детектор по электронному захвату (дэз-детектор)
Он тоже не обязателен по комплектации прибора, но по желанию прибор может быть им укомплектован. В ДЭЗ - детекторе имеется радиоактивный источник, и поэтому приобрести его можно только по специальному разрешению. Он имеет следующее устройство:
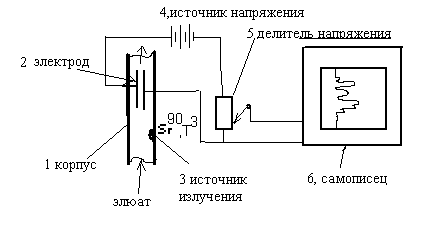
Рис. 5
ДЭЗ-детектор представляет собой трубку, через которую подается элюат, т.е. то, что вышло из хроматографической колонки – газ-носитель и разделенные определяемые вещества. В трубке помещен источник излучения – это стронций либо тритий.
Основным требованием к ДЭЗ-детектору является работа с газом-носителем азотом. При воздействии β-частиц с высокой энергией на атомы азота возникает поток β-частиц с меньшей энергией. Излучение стронция – примерно 1,6 мегаэлектронвольт. В начале выбиваются электроны с энергией сотни тысяч электронвольт. Процесс продолжается дальше. Эти электроны выбивают следующие электроны с меньшей энергией, и так доходит до тепловых электронов, имеющих энергию порядка от 2 – 3 до 10 электроновольт. Тепловые электроны или β-частицы хорошо поглощаются полярными органическими веществами, прежде всего галоидпроизводными, особенно Cl- и Br-производными. Ими поглощаются неполярные вещества, исходя из зарядов на молекулах полярных веществ. Далее, на пути уже поляризованного элюата расположены электроды, к которым приложено электрическое напряжение. Если мало вещества, то все напряжение подается на измерительный прибор, а если много, то оно сжигается так, чтобы сигнал был меньше, и чтобы он хорошо был зарегистрирован самописцем.
При отсутствии определяемых веществ, поглощающих электроны, наблюдается самая высокая электропроводность в цепи, т.е. самая высокая сила тока, так как есть ионизирующее излучение и есть высокая концентрация электронов. А чем больше электронов, тем больше электропроводность. Когда в межэлектродное пространство попадают определяемые вещества, хорошо поглощающие электроны, то концентрация электронов резко падает, сопротивление цепи растет, и сигнал на реостате уменьшается. Самописец регистрирует обычную хроматограмму. Достоинства ДЭЗ-детектора – очень высокая чувствительность к галоидопроизводным, что очень важно при определении многих хлорорганических пестицидов в окружающей среде и целого ряда других веществ. Особенностью ДЭЗ-детектора является то, что он используется в методе реакционной хроматографии, когда определяемые вещества заранее переводят в галоидопроизводные, чтобы потом их определить с наибольшей чувствительностью.
Фазы в газовой хроматографии
Подвижные фазы – это газы-носители. Наибольшее распространение получили водород, гелий и аргон. Основное требование к газам: они должны быть инертны к определяемым веществам и не должны неподвижно связываться неподвижной фазой. Неподвижные фазы представляют собой либо различные адсорбенты: уголь, полимеры различных неорганических материалов, которые работают по механизму адсорбции определяемых веществ на поверхности. В этом случае мы имеем дело с газо-адсорбционной газовой хроматографией.
Эти фазы очень просты и доступны. Существенным недостатком газо-адсорбционной хроматографии является невозможность работы при высоких температурах, а много определяемых веществ переходят в газовую фазу лишь при высоких температурах.
Второй тип фаз – жидкие неподвижные фазы. Это различные вещества, жидкие даже при обычной температуре, и тем более при повышенной, нанесенные на поверхность носителей. Носителями в данном случае служат, чаще всего, адсорбенты. Это различные природные карбонаты кальция, остатки водорослей, молотый кирпич, но наиболее распространены силикагель и полимеры, на поверхность которых наносится жидкая неподвижная фаза. Жидкая неподвижная фаза обычно представляет собой высококипящее органическое вещество, вязкое, малолетучее. Наиболее распространены среди этих веществ динанилфталат или высшие эфиры ортофталевой кислоты: динамил-, диоктил-, дидецилфталаты. Это жидкости, которые кипят при температуре 300–4000 С. Кроме этого, применяют полимерное вещество – полиэтиленгликоль (это жидкость) и различные селиконовые масла, например, полисилаксаны.
Еще применяют жидкие привитые неподвижные фазы. В отличие от фаз, нанесенных за счет просто адсорбции, привитые фазы выдерживают большие температуры. Они не испаряются и механически более устойчивы. А в случае фаз за счет адсорбции при продувании воздухом носитель может скапливаться, отделяться и оголяться, т.е. это нестойкая система. Привитые неподвижные фазы гораздо надежнее. Чаще всего их получают прививкой на селикагеле высших спиртов, т.е. на поверхность селикагеля по ОН-группам вводят высшие спирты (октиловый, дециловый, октодециловый). При этом получаются длинные радикалы, которые играют роль жидкой неподвижной фазы.
Выбор той или иной фазы зависит от природы разделяемых веществ и температуры, которая при их анализе должна поддерживаться. Главное отличие жидких неподвижных фаз от твердых заключается в том, что основным процессом является не их адсорбция на поверхности, а распределение вглубь, т.е. в объем этой фазы. Таким образом, разделяемые вещества, впрочем, как и газ-носитель, могут заходить вглубь неподвижной фазы. Неподвижные фазы всех типов помещаются в хроматографическую колонку длиной до 6 м и диаметром 3–4 мм. Колонка плотно заполняется газом-носителем и готова к работе.
Основная цель хроматограммы–получение количественных характеристик. Поскольку концентрация вещества пропорциональна площади пика, перед нами встает важная задача – опредение площади пика. Большинство приборов выдают хроматограммы на специальной бумаге. Рассмотрим несколько методов определения концентрации веществ по площади пиков.
Первый, наиболее распространенный способ определения площади пика - это метод h x V, т.е. произведение ½ ширины пика на высоту. В основе метода лежит допущение, что пик – это равнобедренный треугольник. Полученная ошибка - 2,6 % связана с этим допущением, т.к. пик имеет закругления. Этот метод является самым распространенным вариантом работы.
Существует еще метод планиметрии, в котором используется прибор планиметр. Им водят по линиям и получают площадь пика, но этот прибор неточен, ошибка составляет 4 %.
Когда нужна большая точность, применяется следующий метод: вырезают пик, взвешивают его, затем по удельному весу (по плотности) бумаги находят площадь пика. Средняя ошибка в данном случае составляет 1,7 %.
В последнее время применяются электронные системы, основанные на интегрировании, в которых детектор соединен с конденсатором, где накапливается заряд, и площадь оценивается с точностью до 0,5 % и даже ниже. Это более точный метод. На таких приборах получают хроматограммы с полной расшифровкой, на которой имеется номер пика, площадь пика, время удерживания и концентрация вещества.
Площадь пика известна, однако, это не концентрация. Для определения концентрации существуют два основных приема:
метод нормировки,
метод внутреннего стандарта.
Метод нормировки выполняется в двух вариантах.
По первому варианту концентрация веществ находится как отношение площади пика к сумме площадей всех пиков на хроматограмме.
Сумма площадей всех пиков приравнивается к 100 % вещества.
В этом методе считается, что для всех веществ один и тот же коэффициент пропорциональности в уравнении:
Ci = K Si
 Таким
образом, предполагают, что все вещества,
которые вышли из хроматографической
колонки, дают пики пропорциональные
их концентрациям. Но это грубое допущение,
справедливо лишь для детекторов по
теплопроводности лишь для полностью
летучих веществ при использовании
водорода или гелия.
Таким
образом, предполагают, что все вещества,
которые вышли из хроматографической
колонки, дают пики пропорциональные
их концентрациям. Но это грубое допущение,
справедливо лишь для детекторов по
теплопроводности лишь для полностью
летучих веществ при использовании
водорода или гелия.
В![]() торой
вариант метода нормировки более строгий.
В этом методе идет расчет по сумме
площадей пиков, но с учетом коэффициентов
пропорциональности.
торой
вариант метода нормировки более строгий.
В этом методе идет расчет по сумме
площадей пиков, но с учетом коэффициентов
пропорциональности.

Площадь каждого пика умножается на коэффициент пропорциональности, связанный с различной чувствительностью детектора к веществам. Заранее находят по стандартной хроматограмме Кi , приняв для одного Кi= 1.
Если есть нелетучие вещества, в этом случае метод даст завышенные результаты, т.к. часть веществ не вышла из колонки. Эти два варианта расчета применимы при работе методом теплопроводности. Для других методов они малопригодны. Дело в том, что большая часть веществ невидима детектором (нет сигнала), и в этом случае на хроматограмме остается ноль. При работе с легкокипящими веществами или для веществ, застревающих в колонке, этот метод не пригоден.
В этом случае применяется универсальный метод - метод внутреннего стандарта. Он основан на том, что готовят стандартную смесь, в которую вносят известное количество стандартного вещества и всех определяемых веществ. Выбирается стандартное вещество, имеющее время удерживания или объем удерживания отличные от этих же параметров веществ в смеси. Снимают стандартную хроматограмму и определяют коэффициенты пропорциональности определяемых веществ по отношению к стандарту. Далее в анализируемую смесь вносят известное количество стандартного вещества и снимают хроматограмму. Концентрация вещества находится по уравнению:
![]()
 Достоинство
метода внутреннего стандарта проявляется,
когда смесь содержит нелетучие, или
летучие, но недетектируемые вещества,
или, когда на хроматограмме есть пики
неизвестных веществ с неизвестными
Кi..
В
качестве стандартных веществ очень
часто выбирают фенол, октан, его замещенные
для алифатических углеводородов и
некоторые другие вещества.
Достоинство
метода внутреннего стандарта проявляется,
когда смесь содержит нелетучие, или
летучие, но недетектируемые вещества,
или, когда на хроматограмме есть пики
неизвестных веществ с неизвестными
Кi..
В
качестве стандартных веществ очень
часто выбирают фенол, октан, его замещенные
для алифатических углеводородов и
некоторые другие вещества.
По предыдущим методам сумма концентраций составляла 100 %, а по последнему методу меньше – 50 или 40 %, т.е. вещества не вышедшие из колонки не исказили концентрацию.
Обычно снимают хроматограмму, смотрят по времени удерживания, какие вещества вышли. Потом подбирают стандарт, вводят его и смотрят, чтобы он попал на пустое место в хроматограмме, чтобы не было перекрывания пиков.
Мы вплотную подошли к теории газовой хроматографии, которая наиболее разработана для газожидкостного варианта. Рассмотрим следующие рисунки:

Рис. 7
а - пик размыт, б – разделен нечетко, в – четкое разделение пиков, г – пики разделены пространством чистого растворителя.
П очему
эти хроматограммы различны? Ведь это
один и тот же прибор и один и тот же
детектор. Ответ на этот вопрос дает
тарелочная
теория газовой хроматографии.
Действительно, в одном случае применяется
колонка с числом теоретических тарелок
2 – 3, во втором – 15 – 20, в третьем – 100,
а в четвертом – 1000 и более. Для того,
чтобы подойти к теории газовой
хроматографии, мы разберем структуру
пика:
очему
эти хроматограммы различны? Ведь это
один и тот же прибор и один и тот же
детектор. Ответ на этот вопрос дает
тарелочная
теория газовой хроматографии.
Действительно, в одном случае применяется
колонка с числом теоретических тарелок
2 – 3, во втором – 15 – 20, в третьем – 100,
а в четвертом – 1000 и более. Для того,
чтобы подойти к теории газовой
хроматографии, мы разберем структуру
пика:
Рис. 8
Где: А - аналитический сигнал или концентрация в зависимости от температуры, времени либо от объема выходящего элюата; tr – время удерживания, Vr – объем удерживания, W- ширина основания пика
Эти величины связаны между собой через скорость газа-носителя F, выраженную в соответствующих единицах. Если объем удерживания выражен в мм, а время удерживания в минутах, то тогда скорость выражается в мм/мин. Очень важная характеристика – это ширина основания пика - ΔW. Пик может быть с широким основанием, а может быть узкий, стрелоподобный. Понятно, что такой пик лучше. А это зависит от многих факторов. Объем связан с коэффициентом распределения вещества и объемами подвижной и неподвижной фазы следующим уравнением:
Vr = D Vs + Vm,
где D ·Vs = V c – исправленный объем удерживания;
И![]() справленный
объем удерживания и просто объем
удерживания различаются на величину
Vm – объем мобильной фазы, который
находится в колонке. Объем мобильной
фазы можно экспериментально определить,
как время выхода веществ, не разделяющихся
колонкой. В верхнюю часть колонки
вводят вещества. В ней они не разделяются.
Чтобы вещества вышли из колонки, надо
чтобы они прошли некоторый путь. Хотя
они не поглощаются, но для них все равно
существует некий объем удерживания,
т.е. время, за которое полностью произойдет
смена газа в колонке. Это объем полостей,
заполненных газом в колонке. Поэтому,
объем удерживания (Vr) классически не
совсем однозначно связан с природой
вещества. Более четко с природой вещества
связан исправленный объем удерживания
(Vc), который более строг и зависит,
например, от качества наполнения
колонки. Величина Vc является очень
важной качественной характеристикой
вещества. В особенности это важно, когда
пиков у хроматограммы много. Когда пиков
2 – 3, то их легко отнести к каким-либо
веществам. Когда пиков 100, то, зная Vm и
даже Vс, их трудно отнести к какому-нибудь
веществу.
справленный
объем удерживания и просто объем
удерживания различаются на величину
Vm – объем мобильной фазы, который
находится в колонке. Объем мобильной
фазы можно экспериментально определить,
как время выхода веществ, не разделяющихся
колонкой. В верхнюю часть колонки
вводят вещества. В ней они не разделяются.
Чтобы вещества вышли из колонки, надо
чтобы они прошли некоторый путь. Хотя
они не поглощаются, но для них все равно
существует некий объем удерживания,
т.е. время, за которое полностью произойдет
смена газа в колонке. Это объем полостей,
заполненных газом в колонке. Поэтому,
объем удерживания (Vr) классически не
совсем однозначно связан с природой
вещества. Более четко с природой вещества
связан исправленный объем удерживания
(Vc), который более строг и зависит,
например, от качества наполнения
колонки. Величина Vc является очень
важной качественной характеристикой
вещества. В особенности это важно, когда
пиков у хроматограммы много. Когда пиков
2 – 3, то их легко отнести к каким-либо
веществам. Когда пиков 100, то, зная Vm и
даже Vс, их трудно отнести к какому-нибудь
веществу.
Следующая характеристика – относительный объем удерживания. В этом случае качественной характеристикой служит не сам исправленный объем удерживания или исправленное время, а отношение времени анализируемого вещества и какого-то стандарта. Причем этот стандарт может быть специально введен в колонку, либо найден. Например, если из 100 веществ какое-то известно точно, то его и выбирают в качестве стандарта и относительно его считают исправленный объем неизвестных пиков:

В![]() ажной
характеристикой является коэффициент
разделения Rs, который количественно
равен отношению двух расстояний между
центрами пиков, деленными на сумму их
оснований:
ажной
характеристикой является коэффициент
разделения Rs, который количественно
равен отношению двух расстояний между
центрами пиков, деленными на сумму их
оснований:
Г рафически
это находится легко. Когда Rs = 1, то площадь
перекрывания пиков составляет не более
10 %. Величина Rs, согласно теории ГЖХ
метода, сложным образом связана с
коэффициентами распределения разделяемых
веществ и новым понятием n – числом
теоретических тарелок:
рафически
это находится легко. Когда Rs = 1, то площадь
перекрывания пиков составляет не более
10 %. Величина Rs, согласно теории ГЖХ
метода, сложным образом связана с
коэффициентами распределения разделяемых
веществ и новым понятием n – числом
теоретических тарелок:

где n – число теоретических тарелок,
Dв и Dа – коэффициенты распределения веществ.
Причем Dв должен быть больше, чем Dа, т.е. числитель должен быть положительным. В формуле имеется еще один член – отношение коэффициента массового распределения вещества к знаменателю 1 + Dmв.. Если Dmв близок к нулю, то и степень разделения в независимости от Dа будет маленькая. Главное в этом уравнении то, что при равенстве коэффициентов распределения Dв и Dа Rs = 0, т.к. числитель превращается в нуль.
Но важнейшая величина это n – число теоретических тарелок колонки. Эта величина принята в соответствии с ректификационным процессом и, примерно, пропорционально числу актов сорбция - десорбция некоторого стандартного разделяемого вещества в
![]()
хроматографической колонке. Чем больше таких актов сорбция – десорбция, тем лучше степень разделения. Число n определяется экспериментально по следующей формуле:
где Vr – исправленный объем удержания.
Число теоретических тарелок будет тем больше, чем меньше основание пика и чем больше будет Vc. Величина n определяется двумя составляющими хроматографичкеской колонки – ее длиной l и высотой, эквивалентной теоретической тарелке h:
![]()
где l – длина колонки, h – ВЭТТ высота, эквивалентная теоретической тарелке, она пропорциональна длине хроматографической колонки, на которой совершается один акт сорбция – десорбция.
Понятно, что n можно увеличивать длиной колонки, а можно снижением h. Величина h может быть рассчитана по уравнению Ван-Деемтера:
г![]() де
F – скорость газа-носителя; В и С –
постоянные члены, зависящие от природы
наполнителя хроматографической колонки,
А – коэффициент, который характеризует
вихревую диффузию, С – коэффициент,
который характеризует диффузию веществ
внутри неподвижной фазы.
де
F – скорость газа-носителя; В и С –
постоянные члены, зависящие от природы
наполнителя хроматографической колонки,
А – коэффициент, который характеризует
вихревую диффузию, С – коэффициент,
который характеризует диффузию веществ
внутри неподвижной фазы.
Все эти коэффициенты, в конечном итоге, описывают динамику разделяемых веществ.
Я хотел бы задать Вам один вопрос: какое минимальное значение h может быть? Минимальное значение h не может быть меньше, чем объем неподвижной фазы в какой-то части колонки, который равен пробе вещества. В идеале, если не будет размывания зоны веществ, то h может быть не меньше, чем длина колонки, в которой помещается объем газовой пробы. Вводим пробу определяемого вещества. Проба имеет какой-то объем, не нулевой, и она будет размещаться вдоль колонки. В верхней части колонки есть разделяемые вещества. Они есть и в нижней части колонки. Они заняли определенный объем, и не может быть меньше. Сконцентрировать их наша система не может, а может только разделить. А эта зона, где есть разделяемые вещества, может только размыться. Размывание этой зоны характеризуется уравнением Ван-Деемтера. Теоретически - это изображение пробы на хроматограмме. Но это в идеальном случае, который почти не достижим. Коэффициент А характеризует размывание зоны, связанное с тем, что между зернами образуются полости. В полостях возможны завихрения, т.е. поток газа не ламинарный, прямолинейный, а с завихрениями. Естественно, что молекулы, которые попали в завихрения, отстали от тех, которые проскочили. Есть другой, обратный эффект – пристеночный. Те молекулы, которые оказались возле стенки, обогнали своих соседей, и это вызывает размывание зон. Так вот, коэффициент А определяется прежде всего размерами зерен хроматографической колонки: чем они мельче, тем меньше пристеночный эффект и тем меньше эти полости, где происходит заметное отставание. Поток получается более стабильным для мелких зерен, поэтому в некоторых видах хроматографии (уравнение Ван-Деемтера относится ко всем видам хроматографии), зерна достигают диаметра одного микрона. Коэффициент В – это молекулярная осевая диффузия. Диффузия – это броунское движение, это то, что молекулы не стоят на месте, а движутся. Они не только успевают переместиться в неподвижную фазу и наоборот, но еще и движутся друг относительно друга. Причем некоторые молекулы во время движения газовой фазы движутся, обгоняя средние, а другие, отставая, и это приводит к размыванию зон. Этот эффект отражен в знаменателе. Чем больше скорость, тем эффект диффузии меньше, понятно почему, и тем меньше времени веществам необходимо, чтобы их молекулы разбежались. Теоретически представьте себе следующую ситуацию: вы ввели в хроматографическую колонку разделяемые вещества и прекратили движение газа. Что произойдет? Через некоторое время молекула распределятся все равномернее по длине колонки. Это эффект молекулярной диффузии. Чем больше скорость, тем меньше влияние этого эффекта на размывание зоны и на увеличение h.
Коэффициент С характеризует движение в неподвижной фазе. Если одна молекула зайдет внутрь неподвижной фазы глубже, а вторая не так глубоко, то, естественно, что это скажется на динамике движения молекул. Зашедшая глубже молекула отстанет, причем этот эффект будет выражен тем больше, чем больше толщина неподвижной фазы. Устраняют этот эффект тем, что неподвижную фазу наносят на поверхность очень тонким слоем – десятая доля микрона, или, как в привитых фазах, создают мономолекулярный слой. В этом случае молекулы не могут застрять надолго в неподвижной фазе. Естественно, что коэффициент С возрастает с ростом скорости подвижной фазы. Получается противоречивая картина: F и в знаменателе, и в числителе. А есть F оптимальное, которое вытекает из графического решения уравнения Ван-Деемтера. Член CF при больших скоростях определяет высоту эквивалентную теоретической тарелке. При небольших скоростях газа-носителя определяют второй член уравнения Ван-Деемтера - ВЭТТ (высоту эквивалентную теоретической тарелке).
Коэффициент В никак не снизить, он определяется температурой. Чем она выше, тем выше молекулярная диффузия. На этом графике имеется минимум, где h – минимальная. Значительный, иногда решающий вклад в величину h вносит член А – вихревая диффузия. Это уравнение отвечает на важнейшие вопрос теории хроматографии - как получить хроматографическую колонку с максимальным числом n.
График уравнения Ван-Деемтера представляет собой повернутую гиперболу. Член CF при больших скоростях определяет высоту эквивалентную теоретической тарелке. При небольших скоростях газа-носителя определяют второй член уравнения Ван-Деемтера ВЭТТ (высоту эквивалентную теоретической тарелке). На этом графике имеется минимум, которому соответствует h – минимальная. В эту величину h значительный (иногда решающий) вклад вносит член А – вихревая диффузия.