

4.2.2.3 Мукополисахаридозы
Недостаточность ферментов лизосом. Ферменты или ферментные системы обычно локализуются в одном определенном районе клетки. Например, ферменты системы транспорта электронов и окислительного фосфорилирования ADP, как и другие ферменты, окисляющие пируват, жирные кислоты и некоторые аминокислоты, локализуются в митохондриях. Многие гидролитические ферменты сконцентрированы в лизосомах [1128]. Если в результате разрушения мембраны лизосомы эти ферменты выходят в цитоплазму, вся клетка подвергается самоперевариванию и гибнет. В норме процесс переваривания осуществляется внутри лизосом, в которых расщепляются не только дефектные компоненты клетки и материал межклеточной соединительной ткани, но и внешний материал, поглощаемый клеткой. К клеточным элементам, которые подвергаются деградации в лизосомах, относятся мукополисахариды, муколипиды и сфинголипиды (рис. 4.8). В настоящее время описаны дефекты многих ферментов, участвующих в деградации [1029, 1240, 1241].
Метаболические пути, нарушения которых вызывают дефект эритроцитов и, следовательно, гемолитическую анемию, уже известны биохимикам, и анализ недостаточности ферментов заключался в изучении отдельных этапов этих реакций. В случае мукополисахаридозов ситуация была совершенно иной. Вначале были изучены наследственные заболевания, и только потом анализ дефектов позволил установить последовательность ферментативных реакций. Вот почему мы начнем с описания этих заболеваний, а затем перейдем к биохимическим нарушениям, дефектам фермент ов и реконструкции нормального метаболического пути. Этот пример показывает, каким образом наследственные заболевания (фактически эксперименты, поставленные самой природой) помогают понять нормальную физиологию и биохимию.
Мукополисахаридозы: клиническая картина. Мукополисахаридозы - группа редких
заболеваний, для которых характерно сочетание многих, зачастую тяжелых симптомов, от нарушений скелета или системы кровообращения до расстройств умственной деятельности. Клинические симптомы являются результатом накопления в разных тканях избыточных сульфатированных полисахаридов.
В табл. 4.5 приведена классификация, а также главные результаты клинических исследований. За исключением мукополисахаридоза II типа (синдром Хантера), все эти дефекты наследуются как аутосомнорецессивные признаки. Клинические симптомы варьируют от сравнительно легких до крайне тяжелых. Описаны две формы
30 4 Действие генов
|
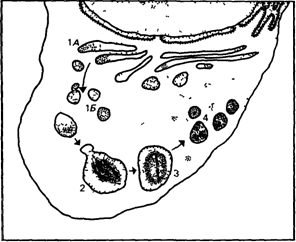
Рис. 4.8. Схематическое изображение функционального цикла нормальной лизосомы. 1А Аппарат Гольджи, 1Б Первичная лизосома, 2 Фаголизосома, 3 Вторичная лизосома, 4 Остаточное тело (Courtesy of Dr Buselmaier) |
синдрома Хантера (30990), которые удается различить у детей старше четырех лет тяжелая «ювенильная» форма, при которой смерть наступает до половой зрелости, и легкая «поздняя» форма, при которой умственная отсталость почти или полностью отсутствует, а прогноз является гораздо более благоприятным Заболевание VI типа (синдром Марото—Лами) (25320) отличается от заболевания I типа (синдром Хурлера) тем, что умственные способности пациентов нормальные и менее выражены аномалии лица В этом случае также наблюдаются два различных подтипа – один с характерным достаточно быстрым течением и тяжелой клинической картиной и другой, прогрессирующий относительно медленно При обоих типах заболевания возможна смерть в возрасте 10-30 лет от нарушений в работе сердца Обнаружены два подтипа IV типа заболевания (синдром Моркио) IVA с тяжелой клинической картиной и IVB, который протекает легче
Отложение запасных веществ в лизосомах и выделение с мочой Гистохимические исследования показали, что эти патологические состояния вызваны нарушениями отложения запасных веществ Во многих клетках, в том числе в фибробластах, клетках печени, клетках Купфера, ретикулярных клетках селезенки и лимфатических узлов, лейкоцитах, эпителиальных клетках почечных клубочков и нервных клетках наблюдаются вакуолизация и увеличение размеров в результате отложения больших количеств запасного материала Установлено, что главными запасными веществами являются сульфатированные гликозаминогликаны
Электронно-микроскопические исследования позволили прояснить ситуацию Было установлено, что запасные соединения откладываются в вакуолях округлой формы, напоминающих лизосомы, которые можно визуализировать у экспериментальных животных после введения им неметаболизируемых соединений Поэтому был сделан вывод, что эти вакуоли представляют собой лизосомы, наполненные непереваренными или лишь частично расщепленными гликозаминогликанами [1129] Это подтвердилось и для других тканей Хотя дефекты метаболизма оставались неисследованными, нарушения отнесли к лизосомным болезням, так как они сопровождались явной перегрузкой системы лизосом Важнейшая функция лизосом заключается в гидролитическом расщеплении макромолекул, поэтому представлялось вероятным, что отложение избыточных запасных веществ связано с недостаточностью гидролитических ферментов лизосом Другим доказательством нарушения метаболизма гликозаминогликанов было избыточное выделение этих соединений с мочой Табл 4 6 дает некоторое
Таблица 4.5. Основные клинические признаки мукополисахаридозов [1029; 1312]
Мукополисахаридоз |
Умственная или двигательная отсталость |
Задержка роста |
Грубые складки на лице |
Дисплазия костей |
Общие контрактуры |
Гепатоспленомегалия |
Помутнение хрусталика |
Характер наследования |
|
Тип 1) |
Название |
||||||||
|
|
||||||||
IH |
Синдром Хурлера (1) |
+ + + |
+ + |
+ + + |
+ + + |
+ + |
+ + |
+ |
Аутосомнорецессивный |
IS |
Синдром Шайе (1) |
– |
– |
± |
+ |
+ |
+ |
+ |
|
IH/S |
Компаунд по синдромам Хурлера и Шайе О) |
+ |
+ |
+ + |
+ + |
+ + |
+ |
+ |
|
IIA |
Тяжелый синдром Хантера (2) |
+ |
+ |
+ |
+ + |
+ |
+ |
+ |
Х-сцепленный, рецессивный |
IIВ |
Легкий синдром Хантера (2) |
+ |
+ |
+ |
+ + |
+ |
+ |
|
|
IIIА |
Синдром Санфилиппо А (ЗА) |
+ + + |
|
+ |
+ |
± |
+ + |
– |
Аутосомнорецессивный |
IIIВ |
Синдром Санфилиппо В(ЗВ) |
+ + + |
– |
+ |
+ |
+ |
+ + |
– |
|
IIIС |
Синдром Санфилиппо С (ЗС) |
+ + + |
– |
+ |
+ |
+ |
+ + |
– |
|
HID |
Синдром Санфилиппо D (3D) |
+ + + |
– |
+ |
+ |
± |
+ + |
– |
|
IVA |
Синдром Моркио А (4) |
– |
++ + |
+ |
+ + + |
+ |
+ |
+ |
Аутосомнорецессивный |
IVB |
Синдром Моркио В (4) |
– |
+ + |
+ |
+ + |
+ |
+ |
+ |
|
V |
|
резервный номер |
|
|
|
|
|
|
|
IVA |
Классическая форма синдрома Марото— —Лами (6) |
– |
+ + |
+ + |
+ + |
+ |
+ |
+ |
Аутосомнорецессивный |
VIB |
Легкая форма синдрома Марото— —Лами (6) |
– |
+ |
+ |
+ |
+ |
+ |
+ |
|
VII |
Скрытый мукополисахаридоз (7) |
+ |
± |
± |
+ |
– |
+ + |
+ |
Аутосомнорецессивный |
" Классификация Мак-Кьюсика (1972) с дополнениями. - отсутствует; + иногда наблюдается; + в легкой форме; + + средней тяжести; + + + в тяжелой форме. В скобках приведены обозначения генетических блоков, соответствующие рис. 4.11 и 4.12. |
|||||||||
32 4. Действие генов
Таблица 4.6. Выделение сульфатированных гликозаминогликанов с мочой при мукополисахаридозах [1029] |
|
Мукополисахаридоз |
Выделяющийся гликозаминогликан |
IH и IS |
Дерматансульфат и гепарансульфат в соотношении 3:1 |
II |
Дерматансульфат и гепарансульфат в соотношении 1:1 |
IIIА, IIIВ, |
Гепарансульфат |
IIIС, IIID |
|
IVA, IVB |
Кератансульфат и хондроитин6-сульфат |
VI |
Дерматансульфат, иногда также некоторое количество гепарансульфата (?) |
VII |
Дерматансульфат и, возможно, гепарансульфат |
представление о выделении разнообразных гликозаминогликанов при различных синдромах. Природа выделяющихся веществ отражает характер метаболических нарушений (табл. 4.7).
Биохимия сульфатированных гликозаминогликанов. Сульфатированные гликозаминогликаны представляют собой сложные гетеросахариды, состоящие из длинных полисахаридных цепей, ковалентно связанных с белковым кором. Полисахаридные цепи дерматансульфата, гепарансульфата, хондроитин-4- и хондроитин-6-сульфата построены из чередующихся остатков глюкуроновой кислоты и сульфатированного гексозамина. Кератансульфат отличается от других гликозаминогликанов тем, что вместо глюкуроновой кислоты содержит остатки галактозы. Полимерные цепи насчитывают до 100 остатков. Они связаны с особым участком в молекуле специфического белка. К одной молекуле полипептида могут быть присоединены несколько полисахаридных цепей. Такие протеогликаны
Таблица 4.7. Дефекты метаболизма при мукополисахаридозах [1029] |
|||
Мукополисахаридоз 1) |
Главное накапливающееся вещество |
Дефектный фермент |
|
IH |
Синдром Хурлера |
Дерматансульфат и гепарансульфат |
α-L-идуронидаза |
IS |
Синдром Шайе |
|
— »— |
IH/S |
Компаунд по синдромам Хурлера и Шайе |
|
-»- |
ПА |
Тяжелая форма синдрома Хантера |
Дерматансульфат и гепарансульфат |
Идуронатсульфатаза |
IIВ |
Легкая форма синдрома Хантера |
-»- |
—» — |
IIIА |
Синдром Санфилиппо А |
Гепарансульфат |
Гепаран-N-сульфатаза |
IIIВ |
Синдром Санфилиппо В |
|
a-N-ацетилглюкозаминидаза |
IIIС |
Синдром Санфилиппо С |
|
а-глюкозаминидаза (?) |
IIID |
Синдром Санфилиппо D |
|
N-ацетилглюкозамин-6-сульфатаза |
|
Синдром Моркио А |
Кератансульфат |
N-ацетилгалактозамин-6-сульфатаза |
IVB |
Синдром Моркио В |
|
Р-галактозидаза |
VIA |
Классическая форма синдрома Марото—Лами |
Дерматансульфат |
М-ацетилгалактозамин-4-сульфатаза (арилсульфатаза В) |
VIB |
Легкая форма синдрома Марото—Лами |
|
— » — |
VII |
Скрытый мукополисахаридоз |
Дерматансульфат и гепарансульфат |
β-глюкуронидаза |
1) Классификация Мак-Кьюсика (1972) с дополнениями. |
|||
4. Действие генов 33
|
Рис. 4.9. Димер L-идуроновой кислоты и N-ацетилгалактозамин-4-сульфата в дерматансульфате. |
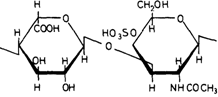
способны образовывать и более крупные комплексы благодаря нековалентным связям. Содержание различных Сахаров в полисахаридных цепях и степень их сульфатирования могут значительно варьировать.
Например, большая часть полисахаридной цепи дерматансульфата построена из повторяющихся димеров Lидуроновой кислоты и N-ацетил-галактозамин-4-сульфата (рис. 4.9). Другие гликозаминогликаны имеют сходное строение. Они являются компонентами соединительной ткани и основного вещества, заполняющего пространство между клетками.
У больных мукополисахаридозами основу структуры гликозаминогликанов соединительной ткани составляют те же крупные протеогликановые образования, которые обнаруживаются и в норме. Отсюда следует, что их синтез не нарушается в результате дефекта фермента. В тканях, где наблюдается аномальное отложение гликозаминогликанов, а также в моче эти молекулы варьируют по длине и имеют меньшие размеры. Это позволяет предполагать, что в клетках происходит расщепление максимально возможного числа связей перед тем, как процесс блокируется из-за дефекта специфического фермента, необходимого для отщепления очередного остатка.
Ферментативные дефекты. Наиболее прямым подходом к исследованию нарушений метаболизма служит поиск соединений, метаболизм которых осуществляется неправильно, и измерение активности ферментов, участвующих в его превращении. Именно таким способом были изучены уже рассмотренные дефекты эритроцитов. Исследования же мукополисахаридозов были затруднены, поскольку не были известны ни детали химической структуры соответствующих гликозаминогликанов, ни ферменты, осуществляющие в норме их катаболизм. Систематическое изучение стало возможным лишь тогда, когда выяснилось, что в культурах фибробластов из кожи больных с синдромами Хантера или Хурлера накапливаются гликозаминогликаны. Важнейшим этапом следует считать доказательство того, что in vitro (в культуре) накопление может быть уменьшено до нормы под действием корректирующего фактора из тканевой жидкости. Впервые это было показано в 1968 г. Неуфельдом и др. [1240]. На основании различий в типе наследования – синдром Хурлера передается потомкам как аутосомнорецессивный признак, а синдром Хантера сцеплен с Х-хромосомой - можно было предположить, что эти заболевания генетически различны, т. е. мутации затрагивают разные реакции расщепления мукополисахаридов. Если бы можно было осуществить слияние ядер клеток, взятых у больного с синдромом Хурлера и у больного с синдромом Хантера (как это было сделано для многих различных типов клеток в работах по генетике соматических клеток), это должно было бы привести к взаимной комплементации дефектов. В ходе решения этой задачи выяснилось, что проблема значительно проще и в слиянии клеток нет необходимости. Для компенсации дефектов, присущих синдромам Хурлера и Хантера, достаточно смешать соответствующие клетки в культуре или даже к клеткам одного генотипа добавить культуральную жидкость, в которой росли клетки другого генотипа. Накопление мукополисахаридов измеряли по включению 35SO4. На рис. 4.10 показаны результаты этих опытов. Компенсация одного из дефектов (синдрома Хурлера) достигалась с использованием как нормальных клеток, так и клеток с другим дефектом (синдромом Хантера) [1086].
В последующие годы подобные эксперименты были выполнены и для других клинических типов, а обнаруженные кор-
34 4, Действие генов
|
Рис. 4.50. Аномальное включение 35SO4 в клетки больных с синдромами Хурлера и Хантера. При смешивании нормальных клеток с клетками больных одним из синдромов включение 35SO4 уменьшается. Такой же результат можно наблюдать при смешивании клеток пациентов, страдающих разными синдромами. Уровень включения при этом близок к норме [1086]. |
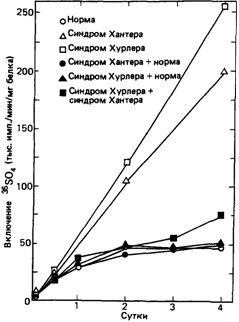
ректирующие факторы охарактеризованы биохимически. Оказалось, что фибробласты больных с синдромом Санфилиппо образуют по меньшей мере две группы, А и В, в которых дефектными являются различные факторы, поэтому клетки групп А и В способны к взаимной коррекции. Таким образом, синдром Санфилиппо генетически неоднороден. С другой стороны, несмотря на глубокие различия в клинической картине синдромов Хурлера и Шайе, фибробласты, взятые у соответствующих больных, дефектны по одному и тому же фактору.
Вскоре было показано, что все корректирующие факторы представляют собой специфические белки. Более подробный анализ показал, что это лизосомные ферменты, участвующие в расщеплении сульфатированных гликозаминогликанов. Важную роль в выяснении механизмов подобных дефектов сыграли и современные представления о структуре этих соединений, и анализ функций корректирующих факторов, и прямые эксперименты по идентификации поврежденных ферментов.
Предполагалось, что при этих нарушениях дефектными являются ферменты, специфически расщепляющие различные типы связей в молекулах гликозаминогликанов. Предсказания на основе этой гипотезы подтверждались экспериментально. Например, при инкубации корректирующего фактора Санфилиппо А (25290) in vitro с гепарансульфатом, выделенным из фибробластов больного с синдромом Санфилиппо и меченным 35SO4, наблюдалось освобождение неорганического сульфата. Дальнейшие исследования показали, что фактор действует на N-сульфатную связь гепарансульфата [1175]. При инкубации in vitro с гепарансульфатом или дерматансульфатом, полученными из фибробластов больного с синдромом Хантера и меченными 35SO4, корректирующий фактор Хантера также катализирует выделение неорганического сульфата. Ген, детерминирующий мукополисахаридоз формы Хантера, сцеплен с Х-хромосомой, поэтому соответствующий дефект фермента должен отличаться от дефекта при синдроме Санфилиппо А. Поскольку общим свойством обоих гликозаминогликанов является случайное распределение сульфатированных остатков идуроновой кислоты, было выдвинуто предположение, что корректирующий фактор Хантера может быть сульфатазой. Это предположение подтвердилось в опытах с искусственным субстратом.
Другой подход к изучению природы ферментативного блока заключается в определении концевых остатков полисахаридных цепей, которые накапливаются при заболевании. Например, было показано, что при синдроме Хантера концевой остаток накапливающегося дерматансульфата представляет собой сульфатированный остаток идуроновой кислоты. Это хорошо согласовывалось с предположением о дефекте сульфатазы, на что указывали опыты с искусственным субстратом. Был сделан вывод, что в норме гликозаминогликаны расщепляются поэтапно и этот процесс
4. Действие генов 35
останавливается, если отсутствует фермент, ответственный за очередной этап. Последовательность моносахаридных остатков в цепи варьирует; такой характер расщепления делает объяснимым тот факт, что накапливающиеся при этих заболеваниях полисахаридные цепи имеют разную длину.
Эти же методы использовали для изучения дефектов других ферментов. Во всех случаях концевые остатки содержали связь, для которой не было соответствующего фермента. Природа ферментативных дефектов позволила объяснить и другое свойство накапливающегося материала: единичное нарушение приводит к накоплению химически неидентичных молекул. Например, у больных с синдромом Хурлера накапливается как дерматансульфат, так и гепарансульфат. Оба соединения содержат остатки a-L-идуроновой кислоты. Поэтому дефект фермента, который строго специфичен в отношении этого остатка, вызывает накопление обоих типов содержащих его полисахаридов.
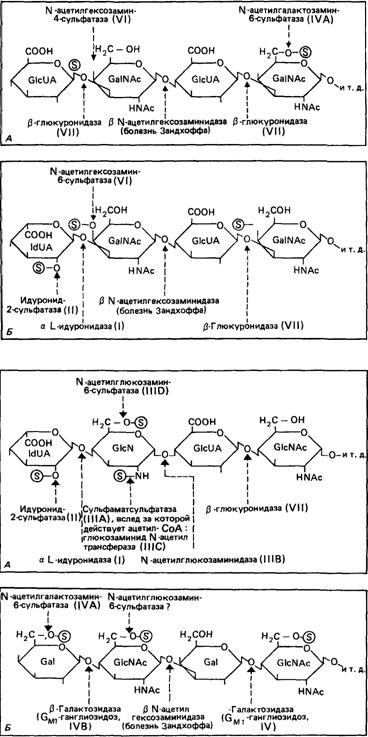
Обобщенные результаты ряда исследований представлены для хондроитина и дерматансульфата на рис. 4.11, а для гепарансульфата и кератансульфата на рис. 4.12. Указаны ферменты, для которых установлены генетические дефекты, перечисленные в табл. 4.7.
Нарушения ферментов и генетическая гетерогенность. В разд. 3.3.5 на примере мышечной дистрофии продемонстрирован анализ генетической гетерогенности с использованием генетических данных (различных типов наследования), а также клинических данных (возраст начала заболевания, характер проявления, тяжесть симптомов и др.). При изучении мукополисахаридозов такой анализ продемонстрировал выраженную межсемейную вариабельность всех этих показателей, между тем внутри каждой семьи проявления были обычно сходными. Поэтому казалось логичным выделить различные генетические типы, что и было сделано еще до исследования ферментативных нарушений. Интересно, как эта классификация, основанная на косвенных данных по изучению фенотипа, согласуется с прямыми данными, полученными при анализе ферментативных нарушений?
В целом соответствие оказалось вполне удовлетворительным (табл. 4.5 и 4.7). Однако имеются два исключения.
1. Согласно клиническим данным, синдром Санфилиппо следует рассматривать как единое заболевание. Было показано, однако, что он может быть обусловлен четырьмя различными ферментативными дефектами. Такая ситуация характерна для многих дефектов метаболизма. Нарушения различных реакций одного и того же пути часто приводят к одинаковой клинической картине. Например, различные дефекты гликолиза вызывают несфероцитарную гемолитическую анемию (разд. 4.2.2.2).
2. С другой стороны, синдромы Хурлера и Шайе сильно различаются: последний протекает значительно легче. Тем не менее оказалось, что в основе обоих заболеваний нарушение одного и того же фермента. Как объяснить этот факт? Весьма возможно, что данный фермент состоит из нескольких полипептидных цепей. Мутации, вызывающие синдромы Хурлера и Шайе, могут затрагивать различные цепи и детерминировать разный уровень остаточной активности фермента. До сих пор, однако, не найдено никаких различий в остаточной активности a-L-идуронидазы в фибробластах больных с синдромами Хурлера и Шайе. Можно предположить и другие механизмы, исходя, например, из результатов генетического анализа гемоглобинов (разд. 4.3).
Изучение генетической гетерогенности мукополисахаридозов еще не проводилось. Однако нет оснований предполагать, что в этом случае различия между мутациями окажутся менее явными, чем те, которые обнаружены для G6PD. Мутантные аллели, которые несет данный, конкретный больной, страдающий аутосомно-рецессивным заболеванием, могут оказаться идентичными. Так бывает, если родители являются родственниками, например двоюродными, или в генетически изолированных популяциях (разд. 6.4.1). Однако в большинстве других случаев мутации, обнаруживаемые у больного,
36 4. Действие генов
|
Рис. 4.11, 4.12. Помимо мукополисахаридозов, рассмотренных в тексте, известны и другие дефекты, приводящие к муколипидозам (болезнь Зандхоффа; ганглиозидоз М II). (По Kresse et al., Klin Wochenschr, p. 870/71, 1981). Рис. 4.11. А. Схематическое изображение структуры и катаболизма хондроитинсульфата. Последовательное расщепление начинается с невосстанавливающего конца (слева). Названия болезней, обусловленных потерей активности ферментов, приведены в скобках. GlcUA, глюкуроновая кислота; GalNAc, N-ацетилгалактозамин; S, SO3H. Б. Схематическое изображение структуры и катаболизма дерматансульфата. Детали см. на рис. 4.11. A. IdUA, идуроновая кислота. Рис. 4.12. А. Схематическое изображение структуры и катаболизма гепарансульфата. Детали см. на рис. 4.11. A. GlcN, глюкозамин; GlcNAc, N-ацетилглюкозамин, ldUA, идуроновая кислота. Б. Схематическое изображение структуры и катаболизма кератансульфата. Детали см. на рис. 4.11. A. Gal, галактоза; GlcNAc, N-ацетилглюкозамин.
|
4. Действие генов 37
имеют различное происхождение, и поэтому маловероятно, что они идентичны. Если термин «гомозигота» использовать строго для обозначения индивидов с абсолютно идентичными мутациями, многих или даже большинство больных рецессивными заболеваниями следует признать негомозиготными. Их можно назвать «составными гетерозиготами» (рис. 3.12). Описан ряд случаев, по клиническим проявлениям промежуточных между синдромами Хурлера и Шайе [1312]. Было показано, что в фибробластах отсутствует корректирующий фактор Хурлера. Некоторые из таких больных действительно могли быть составными гетерозиготами с аллелями Хурлера и Шайе. Однако по меньшей мере в четырех случаях родители больных были родственниками. Возможно существование и третьего аллеля с промежуточным фенотипическим проявлением, поскольку в семьях, где происходит сегрегация двух различных аллелей, заболевание нельзя объяснить последствиями близкородственных браков.
Дифференциальная диагностика и лечение мукополисахаридозов. Если на основании клинических данных подозревается мукополисахаридоз, предполагаемый диагноз должен быть подтвержден обнаружением в моче избыточных сульфатированных гликозаминогликанов. Для этого в настоящее время разработаны соответствующие методы [1312]. Окончательный диагноз, однако, требует выявления дефектного фермента в культуре фибробластов, в лейкоцитах или, в случае некоторых ферментов, в сыворотке [1029]. Если ген экспрессируется в клетках амниотической жидкости, возможна и пренатальная диагностика (разд. 9.1.1). В этом случае либо измеряют накопление радиоактивных гликозаминогликанов, либо определяют природу дефекта фермента. Однако, поскольку количество амниотических клеток, которые удается культивировать, ограниченно, оказывается возможным определить активность лишь нескольких ферментов. Вот почему пренатальной диагностике должно предшествовать скрупулезное энзимологическое исследование больного сибса. Такой предварительный анализ позволяет измерять в клетках амниотической жидкости активность только того фермента, который оказывается дефектным у этого сибса.
Известно, что при определенных обстоятельствах ферменты могут поглощаться дефектными клетками, что приводит к коррекции дефекта. Этот факт свидетельствует о возможности ферментной терапии. Однако до сих пор не удавалось получить достаточно очищенных ферментов, а переливания больших количеств лейкоцитов или сыворотки приводили лишь к незначительному улучшению, в большинстве же случаев результаты были сомнительными. Поглощение клеткой лизосомного фермента является высокоспецифичным процессом, в котором участвует определенный маркерный участок, распознаваемый в полипептидной молекуле фермента; он может быть разным для разных тканей [1240]. Тем не менее этот подход представляется многообещающим.
Дефект маркера для распознавания лизосомных гидролаз [1242]. В 1967 г. Де Марс и Лерой обнаружили «странные» клетки в культуре фибробластов кожи, взятых у
38 4 Действие генов
больного с предполагаемым синдромом Хурлера. Эти клетки имели плотные включения, выявляемые с помощью фазовоконтрастной микроскопии и содержащие кислую фосфатазу. Заболевание назвали I-клеточной болезнью (от английского inclusion-включение). По клиническому проявлению она напоминает синдром Хурлера, но протекает тяжелее и наследуется как аутосомно-рецессивный признак Оказалось, что фибробласты таких больных дефектны по Р-гексозаминидазе, арилсульфатазе А, β-глюкуронидазе, в то же время в культуральной жидкости перечисленные ферменты присутствуют в повышенной концентрации. Чтобы объяснить этот факт, было высказано предположение, что фибробласты больных имеют дефектную клеточную мембрану, однако впоследствии оказалось, что in vitro лизосомы клеток больных поглощают и накапливают нормальные ферменты с нормальной скоростью. В то же время гидролазы из I-клеток не поглощаются нормальными клетками. Следовательно, изменены сами молекулы этих ферментов. Было установлено, что они лишены маркера, необходимого для распознавания при эндоцитозе, т. е. маннозо-6-фосфата. В норме остатки маннозо-6-фосфата присоединяются к ферменту после завершения его синтеза. Они служат сигналом, позволяющим ферменту связываться с рецептором маннозо-6-фосфата, который обеспечивает транспорт лизосомных ферментов в лизосомы, где происходит их активация. В результате нарушения какого-то фермента, участвующего в процессинге лизосомных ферментов, большинство их при I-клеточной болезни лишены маннозо-6-фосфата. Это приводит к тому, что ферменты из клеток секретируются в плазму, а не транспортируются по рецептор-зависимому пути в лизосомы (рис 4 13). Множественные клинические симптомы I-клеточной болезни можно объяснить одним дефектом процессинга, при котором нарушается присоединение к ферментам маркера - маннозо-6-фосфата.
Поскольку участок узнавания является общим для ряда ферментов, I-клеточная болезнь служит примером патологического состояния, при котором дефект одного гена приводит к недостаточности нескольких ферментов. Подобные множественные эффекты могут иметь и другие причины.
|
Рис. 4.13. Схематическое изображение цикла секреции и обратного поглощения гидролитических ферментов лизосом нормальными и мутантными клетками, растущими в культуре. Специфические рспепторные белки, расположенные на поверхности плазматической мембраны всех трех клеток, позволяют им поглощать и транспортировать в лизосомы гидролитические ферменты. Клетки при синдроме Хурлера не способны синтезировать α-L-идуронидазу однако этот дефект можно скомпенсировать добавлением фермента к среде. В случае I-клеточной болезни присут ствуют все ферменты, однако они лишены маркера, необходимого для их распознавания при поглощении и транспорте в лизосомы. |
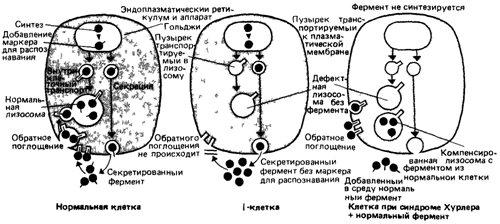
4 Действие генов 39
4.2.2.4 Одновременные нарушения нескольких ферментов
До сих пор во всех случаях - будь то механизм выработки энергии в эритроцитах или катаболизм гликозаминогликанов - рассматривались примеры, при которых одна мутация приводила к изменению или недостаточности единственного фермента. Все это согласуется с гипотезой «один ген - один фермент». Однако известны случаи, когда одна мутация приводит к изменению двух ферментов. Например, активность одного фермента может нарушаться в результате дефекта другого. Так, активность глюкозо-6-фосфатазы при болезни накопления гликогена III типа (23240) уменьшается в результате нарушения амино-1,6-глюкозидазы - фермента, который расщепляет гликоген в точках ветвления молекулы. Изменение структуры фермента представляется маловероятным, поскольку стероиды, обладающие кортизоно-подобным эффектом, вызывают в таких случаях нормализацию активности глюкозо-6-фосфатазы [1199].
Другие случаи, при которых два фермента изменены структурными мутациями, а у гетерозигот их активность примерно в два раза ниже нормы, не удается объяснить таким способом. Вполне возможно, что указанные ферменты имеют общую субъединицу.
|
Рис. 4.14. Аминокислоты с разветвленной боковой цепью. |
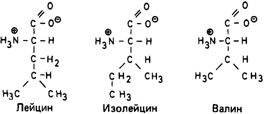
Кетоацидурия, обусловленная дефектом метаболизма аминокислот с разветвленной боковой цепью (болезнь «кленового сиропа») [196]. Известно рецессивное нарушение, которое затрагивает не менее трех функционально родственных ферментов,- болезнь «кленового сиропа». Она вызвана дефектом расщепления аминокислот с разветвленной боковой цепью: лейцина, изолейцина и валина (рис. 4.14). Генетический блок показан на рис. 4.15. При наиболее частой классической форме болезни в течение первой недели после рождения наблю-
|
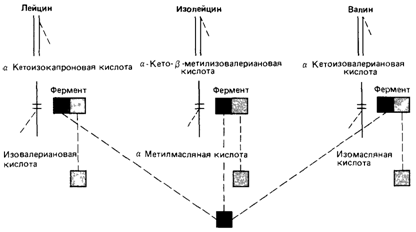
Рис. 4.15. Предполагаемая ферментативная система метаболизма аминокислот с разветвленной боковой цепью содержит общий компонент (А). Эта полипептидная цепь может объединяться с тремя различными полипептидами (В, С, D), образуя три фермента (АВ, АС и AD). Мутационное изменение общей субъединицы приводит к генетическому блоку метаболизма сразу трех аминокислот и вызывает кетоацидурию. Известны редкие случаи дефектов полипептидов В и D. |
40 4 Действие генов
даются затруднение с сосанием, рвота, гипертонус мышц и пронзительные крики. Иногда имеют место потеря тонуса и задержка дыхания (возможно, вследствие гипогликемии). Позже утрачиваются рефлексы, появляются частые припадки, возможна смерть в раннем возрасте. Дети, которые не получали лечения и тем не менее выжили, страдают тяжелой умственной отсталостью [182]. Помимо этого классического типа, описаны «промежуточная», «легкая» и тиамин-зависимая формы болезни.
Анализ генетического блока показал, что активность трансаминаз, которые превращают эти аминокислоты в соответствующие кетокислоты, не изменена (рис. 4.15). Оказалось, что нарушена следующая стадия - окислительное декарбоксилирование. По-видимому, она обеспечивается тремя различными мультиферментными комплексами. Установлено, что эти комплексы имеют один идентичный компонент, который и затрагивает мутация, вызывающая заболевание. При промежуточной форме заболевания наблюдаются периоды атаксии и повышения концентрации аминокислот с разветвленной боковой цепью и соответствующих кетокислот, в особенности при инфекционных заболеваниях. В остальное время показатели крови находятся в пределах нормы, но у больных наблюдаются неврологические отклонения. У одного ребенка при этом заболевании описана остаточная активность фермента, составляющая около 210% нормы. Сходные дефекты описаны в работе [1287].
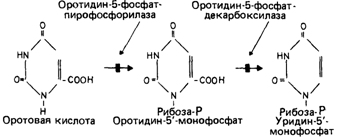
Другие дефекты метаболизма, при которых нарушена активность нескольких ферментов [196]. Другим примером дефекта метаболизма, при котором один генетический блок затрагивает два фермента в одной и той же цепи реакций, служит оротоацидурия (25890), при которой нарушено образование одного из предшественников РНК, уридина, из оротовой кислоты. На рис. 4.16 показаны два генетических блока. У гетерозигот активность обоих ферментов снижена приблизительно вдвое. Это исключает зависимость активности одного фермента от другого и свидетельствует в пользу участия продукта одного и того же гена в обеих реакциях. По меньшей мере в одном случае было установлено, что у гомозиготы второй фермент - декарбоксилаза - имеет измененную электрофоретическую подвижность, а значит, и структуру. Оказалось, что каждая реакция осуществляется одним из двух независимых доменов макромолекулы, которая представляет собой единую полипептидную цепь [1145]. С другой стороны, показано, что при ганглиозидозе Зандхоффа одновременное нарушение активности гексозаминидаз А и В обусловлено мутацией, изменяющей общую для этих ферментов (3-субъединицу [1286].
Описан ряд случаев одновременной недостаточности факторов свертывания крови II, VII, IX и X, зависимых от витамина К. Они вызваны дефектом посттрансляционной модификации [1111b].
Новый взгляд на гипотезу «один ген - один фермент» (или «один ген - одна полипептидная цепь») [1088]. Как уже отмечалось, дефекты единичных генов человека могут нарушать активность сразу нескольких ферментов, что объясняется наличием у этих ферментов общих субъединиц. Это структурные мутации.
|
Рис. 4.16. Два генетических блока двух последовательных этапов биосинтеза пиримидинового основания урацила. Оба блока проявляются как оротоацидурия. |
4. Действие генов 41
Другой важный аспект синтеза ферментов связан с посттрансляционным процессингом. Например, сахарозоизомальтаза построена из двух полипептидных цепей, каждая из которых обладает ферментативной активностью. Эти полипептиды образуются из единого предшественника в результате протеолитического расщепления [1297]. Для образования активного инсулина также необходим процессинг проинсулина.
Весьма убедительные доказательства процессинга пептидов получены для ряда мелких пептидов, синтезирующихся в мозге, - энкефалинов и эндорфинов. В этом случае одна и та же молекула предшественника в результате процессинга может расщепляться на различные пептиды в зависимости от типа клеток или стадии их развития, что может служить одним из механизмов дифференцировки при эмбриональном развитии. На рис. 4.17 показана молекула препродинорфина и ее процессинг с образованием различных типов энкефалинов, эндорфинов и динорфинов. Функции этих белков будут кратко рассмотрены в гл. 8. Подобный посттрансляционный процессинг описан для других гормонов и нейропептидов, синтезирующихся главным образом в гипофизе и гипоталамусе.
Модификации, важные для функционирования молекул, происходят не только на посттрансляционном уровне, но и на уровне экспрессии гена; они могут изменить даже сам ген. Подобная модификация установлена для генов иммуноглобулинов (разд. 4.4). Однако эти факты не умаляют эвристической ценности гипотезы «один ген - один фермент», которая в большинстве случаев верна.
Нарушение работы сразу нескольких ферментов в результате одной мутации может наблюдаться в особых случаях, например если нарушено поглощение, транспорт или связывание кофакторов.