

4.3.4. Талассемии [31; 972; 138; 1253; 222; 97а]
Генетически детерминированное снижение или полное подавление синтеза той или иной цепи гемоглобина обусловливает ряд разнообразных патологических состояний, известных под общим названием «талассемии». Это слово происходит от греческого «Таласса» - Средиземное море, первоначально оно отражало средиземноморское происхождение большинства индивидов - носителей генов, ответственных за талассемию. Хотя с точки зрения географии или этнографии этот термин нельзя считать правильным, он используется достаточно широко. Все случаи талассемии можно под-
Таблица4.17. Мутации, приводящие к β-талассемии [972; 1253] |
|||
Локализация |
Изменение в последовательности |
Тип талассемии |
Этническая группа |
1. Мутации, нарушающие транскрипцию |
|
|
|
а) удаленные регуляторные элементы |
|
|
|
–87 |
C–G |
β+ |
Жители Средиземноморья |
–88 |
С–Т |
β+ |
Негры США |
б) ТАТА-последовательность |
|
|
|
–28 |
А–С |
β+ |
Курды |
–28 |
A–G |
β+ |
Китайцы |
–29 |
A–G |
β+ |
Негры США |
2. Нарушения расщепления РНК на 1000 т. п. н. дальше экзона 3 |
Т–С |
β+ |
Негры США |
3. Неактивная РНК |
|
|
|
а) нарушения терминации |
|
|
|
кодон 17 |
А–Т |
β° |
Китайцы |
кодон 39 |
С–T |
β° |
Жители Средиземноморья |
кодон 15 |
G–A |
β° |
Индийцы |
б) мутации сдвига рамки |
|
|
|
кодон 8 |
–2 |
β ° |
Турки |
кодон 16 |
–1 |
β ° |
Индийцы |
кодон 44 |
–1 |
β ° |
Курды |
кодон 8/9 |
+1 |
β ° |
Индийцы |
кодон 41/42 |
–4 |
β ° |
Индийцы |
кодон 6 |
–1 |
β ° |
Жители Средиземноморья |
кодон 71/72 |
+ 1 |
β ° |
Китайцы |
4. Мутации, затрагивающие процессинг РНК |
|
|
|
а) границы сплайсинга |
|
|
|
донорные участки |
|
|
|
IVS-1, поз. 1 |
G–A |
β ° |
Жители Средиземноморья |
IVS-1, поз. 1 |
G–T |
β ° |
Индийцы |
IVS-2, поз. 1 |
G–A |
β ° |
Жители Средиземноморья |
IVS-1, поз. 5 |
G–C |
β + |
Индийцы |
IVS-1, поз. 6 |
Т–С |
β + |
Жители Средиземноморья |
акцепторные участки |
|
|
|
1VS-1 |
деления 25 п. н. |
β ° |
Индийцы |
IVS-1 |
A–G |
β ° |
Негры США |
б) образование новых сайтов сплайсинга |
|
|
|
новый донорный сайт |
|
|
|
IVS-1, |
С–T |
β° |
Китайцы |
IVS-2, поз. 705 |
T–G |
β+ |
Жители Средиземноморья |
IVS-1, поз. 745 |
C–G |
β+ |
» |
1VS-1, поз. 116 |
T–G |
β+ |
» |
новый акцепторный сайт |
|
|
|
IVS-1, поз. 110 |
G–A |
β+ |
» |
в) усиление криптических сайтов |
|
|
|
кодон 24 |
Т–A |
β+ |
Негры США |
кодон 26 |
G–A |
βЕ |
Жители Азии |
кодон 27 |
G–T |
βKnossos |
Жители Средиземноморья |
90 4. Действие генов
разделить на α- и β-талассемии. Талассемии различаются по этиологии, поскольку показано, что ослабление синтеза цепей гемоглобина может быть обусловлено несколькими генетическими механизмами [1037].
Успехи в исследовании талассемий на молекулярном уровне привели к тому, что мутационные повреждения, характерные для этой группы заболеваний, изучены в настоящее время гораздо полнее любых других мутаций у млекопитающих. Изучение различных генов, ответственных за талассемию, позволило многое узнать о структуре, функции и организации глобиновых генов в норме. Выяснилось, что мутации, затрагивающие различные этапы синтеза гемоглобина, могут ослаблять синтез гемоглобина (β+-талассемий) или даже полностью его предотвращать (β0-талассемии) [972; 1253; 1238; 4341] (табл. 4.17, 4.18).
Транскрипционные или промоторные мутации. Мутации, которые вызывают талассемию и затрагивают 5'-некодирующую область гена Нbβ, можно рассматривать как регуляторые мутации, влияющие на транскрипцию. Такие мутации обнаружены в константном регуляторном элементе с последовательностью PuCPuCCC и внутри ТАТА-последовательности (табл. 4.17). Эти мутации ослабляют синтез гемоглобина и проявляются как относительно легкие формы талассемий. Мутации, затрагивающие СААТ-сайт, в настоящее время не обнаружены.
Мутации в сайте полиаденилирования РНК. У негров часто обнаруживается одиночная мутационная замена ААТААА ААСААА в 3-фланкирующей области гена Нbβ, приводящая к β+-талассемий; таким образом, мутации в 3-некодирующей области также могут влиять на эффективность транскрипции. Относительно слабое проявление β-талассемий в этой расовой группе объясняется явным преобладанием мутаций, затрагивающих ТАТА-последовательность (см. выше) и сайт полиаденилирования (табл. 4.17).
Нонсенс-мутации и мутации со сдвигом рамки считывания. Как уже указывалось ранее, мутации, приводящие к возникновению терминирующего кодона внутри экзона гена гемоглобина, обусловливают синтез укороченной, неактивной цепи и вызывают поэтому β0-талассемию. Выявлены три такие мутации, одна из них характерна для уроженцев Средиземноморья (β39С—Т). Обнаружена рестриктаза (Mael), узнающая
|
Рис. 4.52. Транскрипция и трансляция генов гемоглобина. Номера указывают сайты, которые изменяются при мутациях, вызывающих талассемию. |
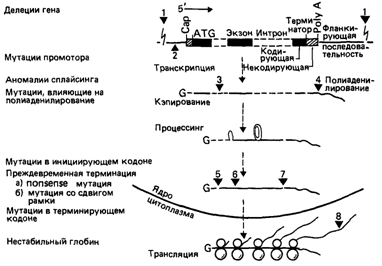
4. Действие генов 91
этот сайт, благодаря чему стала возможной пренатальная диагностика β39-талассемии [1328а].
Делеции и инсерции, длина которых не кратна трем нуклеотидам, нарушают нормальное считывание генетической информации, в результате чего синтезируются нефункциональные цепи глобина. В различных популяциях обнаружено семь подобных мутаций, вызывающих β°-талассемию (табл. 4.17).
Мутации, нарушающие процессинг РНК. Процессинг РНК заключается в вырезании интронов и сплайсинге (сращивании) экзонов с образованием функциональной мРНК (рис. 4.40, 4.52). Описано много различных мутаций, нарушающих этот процесс. Например, известна целая группа мутаций, изменяющих динуклеотид GT (или AG) в донорном (или акцепторном) участке сплайсинга. Эти динуклеотиды входят в состав так называемых канонических (обобщенных) последовательностей, которые включают еще несколько нуклеотидов и играют центральную роль в нормальном сплайсинге. В результате одиночных замен в этих сайтах сплайсинг нарушается, что приводит к β°- или β+-талассемии. В некоторых случаях мутации активируют так называемые криптические сайты с динуклеотидами GT и AG. В норме, вероятно, эти сайты в сплайсинге не участвуют. В результате образование нормальной мРНК нарушается. Мутации в интронах могут генерировать новые сайты сплайсинга в интронах, которые конкурируют с нормальными сайтами, замедляя процессинг, и в конечном счете приводят к талассемии. Мутации другого класса усиливают уже существующие криптические сайты. Часто встречающаяся мутация такого типа НbЕ активирует такой сайт. Это приводит к ослаблению синтеза гемоглобина HbβE и, следовательно, к талассемии. Различные мутации, нарушающие сплайсинг, перечислены в табл. 4.17. Все точковые мутации в гене β-глобина, которые вызывают β-талассемию, показаны на рис. 4.53.
Делеции в β-глобиновом кластере генов и наследственная персистенция фетального гемоглобина. В отличие от а-талассемии Р-талассемия обычно обусловлена не делениями генов. Однако из этого правила есть много исключений. Более трети случаев Р-талассемии у индийцев оказывается связанной с делецией длиной 619 п. н., которая начинается во втором интроне и заканчивается за 3'-концом некодирующей области гена Нbβ (рис. 4.54, табл. 4.18). Различные редкие делеции в этом гене описаны у негров США, известен один случай среди датчан. Обнаружено также несколько других, более крупных делеции в γ-δ-β-локусе. Их локализация и протяженность показаны на рис. 4.54. Методами цитогенетики эти делеции обнаружить не удается: они слишком малы для микроскопического изучения.
Мутации «гемоглобин Lepore» и «гемоглобин Kenya» представляют собой делеции, при которых сохраняются части функциональных генов и возникают их слияния: δ-β (гемоглобин Lepore) и γ-β (гемоглобин Kenya). Известно несколько делеции, при которых происходит утрата всего или почти всего кластера, при этом γ-, δ- и β-цепи не синтезируются. Принято различать делеции, приводящие к талассемическому фенотипу анемии, например δ-β-талассемия, и делеции, при которых отсутствие δ- и β-локусов компенсируется синтезом фетального гемоглобина (наследственная персистенция фетального гемоглобина, НПФГ). Это отличие не является абсолютным, поскольку при НПФГ компенсация благодаря синтезу γ-цепи не бывает полной. Остается
|
Рис. 4.53. Расположение мутаций, вызывающих р-талассемию в гене Нbβ. Различные мутации перечислены в таблице 4.17. |
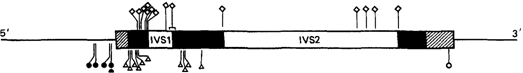
92 4. Действие генов
|
Рис. 4.54. Делеции в кластере генов γδβ. Большинство этих делеций встречается редко. НПФГ (англ. HPFH) - наследственная персистенция фетального гемоглобина. |
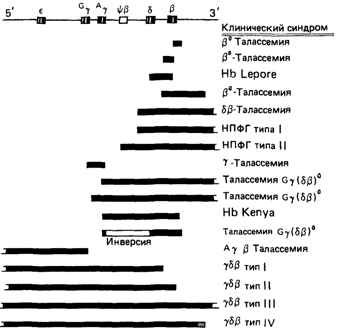
неясным, почему некоторые делеции активируют ген фетального гемоглобина. В настоящее время этот вопрос интенсивно изучается.
Наследственная персистенция фетального гемоглобина может быть вызвана мутациями неделеционной природы. В случае греческой разновидности НПФГ обнаружена точковая мутация в 5'-фланкирующей области Аγ гена в положении —117 [1040]. Другая точковая мутация в положении 202 5'-фланкирующей области гена HbGγ найдена у негров с неделеционной НПФГ [1093]. Считается, что последовательности, которые изменяются под действием этих мутаций, в норме играют ключевую роль в остановке синтеза у-цепи в постнатальный период.
Изучение генетического контроля синтеза гемоглобина имеет большое значение для лечения талассемии и серповидноклеточной анемии, так как усиленный синтез гемоглобина HbF при этих заболеваниях мог бы обеспечить значительный терапевтический эффект.
Гетероклеточная наследственная персистенция фетального гемоглобина [222]. Группа гетерогенных генетических состояний ха рактеризуется некоторым усилением синте за фетального гемоглобина (2-3%, иногд; более) и явно неравномерным распределе нием в популяции эритроцитов. Этим и
Таблица.4.18. β-талассемии, наиболее часто встречающиеся у различных этнических групп [972] |
|||
Этническая группа |
Мутация, приводящая к β-талассемии |
Тип |
Частота, % |
Негры США |
ТАТА-последовательность (-29) |
β+ |
39 |
|
Сайт полиаденилирования |
β+ |
26 |
Жители |
Интрон 1 (поз 110) |
β+ |
35 |
Средиземноморья |
β39-терминатор |
β+ |
27 |
Индийцы |
Интрон 1 (поз. 5) |
β+ |
36 |
|
Делеция (619 п. н.) |
β° |
36 |
Китайцы |
Сдвиг рамки (поз. 71/72) |
β° |
49 |
|
Интрон 2 (поз. 654) |
β° |
38 |
4. Действие генов 93
объясняется название состояния - гетероклеточная НПФГ. Оно не сопровождается анемией. Если ген, определяющий гетероклеточную НПФГ, сочетается в генотипе с геном HbS или с геном β-талассемии, уровень фетального гемоглобина может превышать обычные 2-3%. В частности, при серповидноклеточной анемии повышенный уровень гемоглобина HbF приводит к смягчению клинической картины заболевания. Гетероклеточная НПФГ наследуется как доминантный аутосомный признак и, судя по рестрикционному картированию, не связана с заметными делециями в β-глобиновом кластере. Молекулярные основы гетероклеточной НПФГ неизвестны, но некоторые данные свидетельствуют о том, что геи, ответственный за повышенный уровень гемоглобина F, не сцеплен с γ-δ-β-кластером [1012].
Значение в клинике. β-Талассемии широко распространены в тропических и субтропических областях, поскольку больные, как оказалось, имеют некоторое селективное преимущество перед здоровыми в отношении тропической малярии [1227] (разд. 6.2.1.6). У гетерозигот по β-талассемии наблюдается небольшая анемия (табл. 4.19), несколько повышено количество гемоглобина А2(α2δ2). Их эритроциты мельче и содержат меньше гемоглобина (показатели МСН и MCV снижены) [31; 222]. Гетерозиготы обычно не нуждаются в медицинском наблюдении или лечении. Внешний вид эритроцитов показан на рис. 4.55.
Гомозиготные больные с β-талассемией страдают сильной анемией и нуждаются в переливаниях крови. У гомозигот по β°-талассемии гемоглобин А полностью отсутствует, при β+-талассемии его уровень сильно снижен. Большая часть гемоглобина - это гемоглобин фетального типа. Заболевание характеризуется задержкой роста и приводит к летальному исходу в детстве или юности. Гомозиготы и гетерозиготы-компаунды по β+/β°-талассемии страдают тяжелой гемоглобинопатией. Показано, что одновременное поражение а-талассемией смягчает клиническую картину у гомозигот по р-талассемии.
Сочетание гемоглобина S с β+-талассемией характерно для негритянских популяций и по симптоматике напоминает серповидноклеточную анемию. Сочетание гемоглобина Е с р-талассемией часто встречается в Юго-Восточной Азии и, подобно β°-талассемии, вызывает тяжелую анемию. Это связано с тем, что мутация гемоглобина Е сама по себе приводит к легкой талассемии (см. выше).
К настоящему времени идентифицировано более 30 различных точковых мутаций, обусловливающих β-талассемию (рис. 4.53, табл. 4.17). Из них 33%-нонсенс-мутации и мутации со сдвигом рамки считывания - препятствуют трансляции мРНК, 47% нарушают сплайсинг, 16%-транскрипцию и 1 % - механизм полиаденилирования. По-видимому, известна лишь небольшая часть мутаций, вызывающих талассемию.
Удивительная гетерогенность мутаций в β-глобиновом локусе проявляется и в высокой частоте гетерозигот-компаундов по р-талассемии. Такие пациенты несут сразу две мутации, обусловливающие р-талассемию, и наследуют их от обоих родителей. В изолированных популяциях гетерозиготыкомпаунды встречаются несколько реже, поскольку большинство случаев заболевания в таких изолятах бывает связано с единичными, распространившимися мутациями. Например, нонсенс-мутация β39 составляет приблизительно 27% всех мутаций, вызывающих р-талассемию в среднем по Средиземноморью (табл. 4.18), и в то же время составляет абсолютное большинство на острове Сардиния. Поскольку нарушения у гомозигот могут быть самыми разными (от едва заметных до полного отсутствия синтеза глобина) и гетерозиготыкомпаунды встречаются часто, тяжесть заболевания сильно варьирует. В табл. 4.19 приведены сведения о наиболее важных с клинической точки зрения гемоглобинопатиях.
α-Талассемии делеционной природы [1122, 1156а, 2322]. Большинство случаев α-талассемии связано с делециями генов. В ходе эволюции происходили конверсия генов (разд. 2.3.4) и множественные акты рекомбинации («согласованная эволюция»), что привело к высокой степени гомологии в
94 4. Действие генов
Таблица 4.19. Гемоглобинопатии, имеющие клиническое значение |
|

структурных и фланкирующих областях нормальных генов α-глобинов. Гомология последовательностей в этих областях заставляет хромосомы неправильно спариваться, и при рекомбинации неидентичных спаренных хромосом возникают дупликации и делеции (рис. 4.56). Обнаружены хромосомы, содержащие только один ген (α) или три гена (ααα). Селекция на устойчивость к малярии привела к увеличению в тропических и субтропических популяциях частоты гена Нbα( — α). Это один из наиболее часто встречающихся типов талассемии. Хромосомы с утроенным локусом (ααα), по-видимому, практически не вызывают отрицательных эффектов у их носите-
4 Действие генов 95
|
Рис. 4.55. Мазки крови из периферических сосудов нормального человека (А) и больных гетерозиготного по β-талассемии (Б), гетерозиготного по α-thal-1 (В) и больного тяжелой формой β-талассемии (Г) |

лей и встречаются в этих популяциях гораздо реже
Два типа делеций обусловливают умеренную форму α-талассемии (α-thal или — α /αα) Механизмы делеций проиллюстрированы на рис 4 56 При так называемом левостороннем кроссинговере возникает хромосома с одиночным геном Нbα вследствие ошибочного спаривания последовательностей, расположенных на 5'-конце aj-псевдогена, и гомологичной ей последовательности в локусе Hba2 При рекомбинации происходит утрата участка ДНК длиной 4,7 т п н Поскольку область, в которой происходит рекомбинация в данном случае, расположена перед участком правосторонних делеций (см ниже), используется термин «левосторонний» Так называемый «правосторонний» кроссинговер, являющийся результатом неправильного спаривания между генами Hba2 и Hbαl, приводит к образованию комбинированного гена α2-α1 и делеций в 3,7 т п н Этот «правый» α-ген (точнее, ген α1-α1) – наиболее частая причина α-талассемии в Африке и в Средиземноморье, тогда как в Азии обнаруживаются продукты и левостороннего, и правостороннего кроссинговера Делеция одного гена α в обоих типах мутаций вызывает стертую форму талассемии и очень широко распространена во всем мире, причем частота гетерозигот достигает 33% в некоторых областях Африки и Средиземноморья
Делеций, обусловливающие утрату обоих α-локусов (– –), показаны на рис 4 57
Возникающая в этом случае талассемия часто обозначается как α-thal-1 Такие мутации распространены в Юго-Восточной Азии, редки в Средиземноморье и никогда не обнаруживаются у африканцев
|
Рис. 4.56. Кроссинговер в X и Z участках гомологии в области гена НЬа При «левостороннем» кроссинговере происходит ошибочное спаривание X участков гомологии с последующей рекомбинацией, при которой возникает один ген Нbα (с делецией длиной 4 2 т п н) При «правостороннем» кроссинговере происходит ошибочное спаривание Z гомологичных участков, кроссинговер внутри а-генов приводит к образованию составного гена α2α1 и делеции длиной 3,7 т.п.н В результате обоих событий возникают хромо сомы с тремя α-глобиновыми генами [1156а] |

96 4. Действие генов
|
Рис. 4.57. Делеции в гене Hbα. Большая часть делеций встречается часто (см. текст). ■; ▥ протяженность делеций неизвестна. |
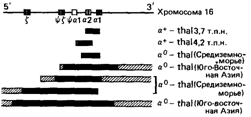
Все делеции можно выявить как до, так и после рождения с помощью рестрикционного картирования. Описаны различные фенотипы, наблюдающиеся при делециях одного, двух, трех и четырех генов а (табл. 4.20, А, рис. 4.57). Отсутствие одного гена НЬа (— α/αα) не приводит к заметным гематологическим нарушениям, поскольку три остальных гена остаются активными. Для обнаружения талассемии при делеции одного из генов Нbα необходим рестрикционный анализ или количественный анализ биосинтеза α-цепи глобина. Делеция всех
четырех α генов (– –/– –) приводит к летальному исходу до или во время родов и известна под названием водянки плода, поскольку наблюдается сильный отек у мертворожденных детей. Большая часть молекул гемоглобина у таких детей состоит из четырех α-цепей (гемоглобин α4 или гемоглобин Bart). Считается, что плод способен доживать до поздних сроков беременности благодаря присутствию функционального гемоглобина Portland (ζ2у2). У африканцев водянка плода практически не встречается, это связано с отсутствием в популяции хромосом, несущих делецию обоих генов α-глобина.
Делеция двух генов α (– –/α+ или – –/сих) приводит к слабовыраженной анемии, тогда как делеция трех генов (—α/ — ) – к сильной анемии, для которой характерно образование гемоглобина НbН – тетрамера β4 (табл. 4.20, А). Гемоглобин НbН образуется вследствие недостатка α-глобина на фоне нормального синтеза β-глобина.
Неделеционная α-талассемия [972, 1156]. Логично предположить, что среди мутаций неделеционной природы имеются такие, которые обусловливают α-талассемию. Действительно, обнаружен широкий спектр таких мутаций (табл. 4.20, Б).
Таблица 4.20, А. α-Талассемии, обусловленные делециями |
||||||
|
Число генов Нbα |
Общее число активных генов Нbα |
Общее число делегированных генов Нbα |
|||
|
материнских |
Отцовских |
||||
|
активных |
делетиро-ванных |
активных |
делегированных |
|
|
Норма |
2 |
— |
2 |
— |
4 |
— |
Слабовыраженная α-талас-семия |
1 |
1 |
2 |
— |
3 |
1 |
(α-thal-2) |
2 |
— |
1 |
1 |
|
|
Тяжелая α-талассемия |
а) – |
2 |
2 |
— |
|
|
(α-thal-1) |
б) 2 |
— |
— |
2 |
2 |
2 |
|
в) 1 |
1 |
1 |
1 |
|
|
Болезнь |
|
|
|
|
|
|
НbН |
— |
2 |
1 |
1 |
1 |
3 |
(НbН = Р4) |
1 |
1 |
— |
2 |
|
|
Водянка плода при Нbγ4 |
— |
2 |
— |
2 |
— |
4 |
Следует отметить, что делеции могут быть унаследованы как от матери, так и от отца или (кроме мягкой формы а-талассемии) от обоих родителей. Кроме того, при различных комбинациях может возникать один и тот же фенотип тяжелой α-талассемии: а) мать: тяжелая α-thal, отец: норма; б) мать: норма; отец: тяжелая α-thal; б) отец: слабая α-thal, мать: слабая α-thal. |
||||||
4. Действие генов 97
Таблица 4.20, Б. Мутации неделеционной природы, вызывающие α-талассемию [972; 1156] |
|

Приведенный в таблице перечень не содержит регуляторных мутаций, локализующихся перед геном Нbα, однако найдена мутация, нарушающая стартовый кодон AUG. Фенотипически это изменение проявляется как α-талассемия – не образуется функциональной мРНК, поскольку AUGкодон, с которого начинается трансляция, находится в 32-м положении. Это приводит к синтезу явно дефектного α-глобина, лишенного первых 32 аминокислот. Известна также мутация в 3'-фланкирующей области, вызывающая снижение синтеза функциональной мРНК.
В настоящее время обнаружена лишь одна мутация, затрагивающая процесс
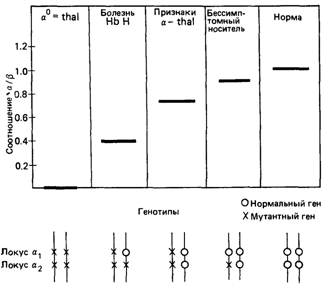
Рис. 4.58. Синтез цепей глобина при α-талассемиях. Синтез молекул глобина представлен в виде отношения Hbα/Hbβ, которое в норме близко к 1, другими словами, α- и β-глобин синтезируются в эквивалентных количествах. Носители, у которых признаки заболевания не выражены, являются гетерозиготами по α-thal-2 (стертая форма α-талассемии), это удается выявить только специальными биохимическими тестами [1159]. |
|
98 4 Действие генов
сплайсинга. Это короткая делеция длиной 5 п.н., удаляющая акцепторный сайт сплайсинга в первом интроне.
Известны четыре различные мутации, изменяющие терминирующий кодон UAA на смысловой. В результате этого образуется цепь α-глобина, удлиненная на 31 аминокислоту по сравнению с нормальной. мРНК, кодирующие такой удлиненный гемоглобин, нестабильны. В крови обнаруживается лишь до 5% мутантного белка. Наиболее часто из этих мутаций встречается гемоглобин Constant Spring (рис. 4.58).
В случае гемоглобина Quong-Sze мутация нарушает способность к образованию димера α1β1 и приводит к сильной нестабильности молекулы. Вероятно, будут обнаружены и другие варианты с подобными свойствами.
Прямых данных о частоте неделеционной α-талассемии нет, однако была изучена природа генов, аллельных генам а-талассемии, у больных с гемоглобином НbН (β4), у которых в одной из хромосом делетированы по меньшей мере два гена Нbα (—/αα). В популяциях Саудовской Аравии и Китая при гемоглобинопатии НbН до 50% таких аллельных генов а-талассемии не содержали делеций.