

4.12. Вплив температури на швидкість біохімічних реакцій
Швидкість хімічних реакцій значною мірою залежить від температури. У загальному вигляді швидкість реакції можна записати таким чином:
v = k(T)cm, (4.84)
де k(T) – константа швидкості реакції, яка залежить від температури; c – концентрація; m – порядок реакції. Проаналізувавши експериментальні температурні залежності, Я. Вант-Гофф, а пізніше С. Арреніус (1889) запропонували емпіричну залежність швидкості реакції від температури:
![]() (4.85)
(4.85)
де Ea – енергія активації; А – передекспоненційний множник, який відображає ймовірність перебігу реакції. Рівняння (4.85) називається рівнянням Арреніуса. Енергія активації Еа – це енергетичний бар’єр, який переборюють молекули для здійснення реакції (рис. 4.34).
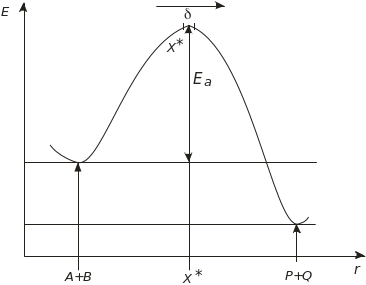
Рис. 4.34. Енергетична крива хімічної реакції:
Е – енергія; Еа – енергія активації; r – координата реакції; Х – активований комплекс. Передекспоненційний множник А – це частота зіткнень реагуючих молекул
Тоді
експоненційний множник
![]() (множник Больцмана) визначає частку
ефективних зіткнень співударів,
які приводять до хімічної реакції.
(множник Больцмана) визначає частку
ефективних зіткнень співударів,
які приводять до хімічної реакції.

Рис. 4.35. Графік Арреніуса:
k – постійна швидкості реакції; Т – температура; - кут нахилу прямої
Для визначення Еа експериментальні дані подаємо у координатах Арреніуса (рис. 4.35):
![]() . (4.86)
. (4.86)
Отримуємо
лінійну залежність
![]() .
З нахилу прямої знаходимо Еа:
.
З нахилу прямої знаходимо Еа:
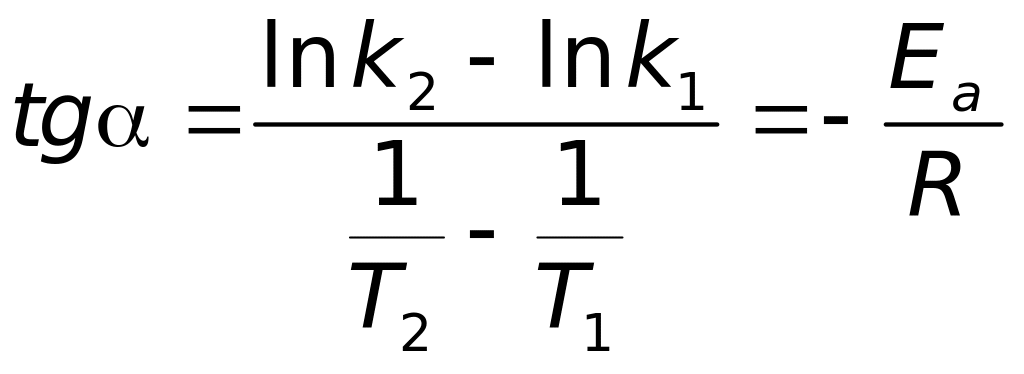 . (4.87)
. (4.87)
Перше пояснення температурної залежності швидкості реакції було зроблено на базі теорії співударів молекул у газовій фазі (В. Мак-Люіс, 1910). Пізніше було розроблено теорію абсолютних швидкостей реакцій (теорію перехідного стану) (Г. Ейрінг, 1935), яка пояснює хімічні реакції й ферментативні реакції в розчині. Ця теорія зв’язує швидкість реакції з термодинамічними характеристиками реагуючих молекул. Теорія абсолютних швидкостей реакцій на відміну від теорії співударів розглядає хімічну реакцію як складний багатоступеневий процес взаємодії ядер та їхніх електронних оболонок, а не просто співударів двох “кульок”. За теорією абсолютних швидкостей реакцій, перехід реагуючих молекул у продукти реакції відбувається через проміжний стан зі збільшеною енергією, який називається активованим комплексом. Утворення такого комплексу лімітує швидкість проходження будь-якої хімічної реакції. Утворення й розпад активованого комплексу зручно описувати через залежність потенційної енергії Е реагуючих молекул від координати реакції r (рис. 4.34). Координата реакції визначає таку траєкторію перебігу реакції, коли елементарні переходи реагуючих молекул від одного стану до іншого відбуваються з мінімальними енергетичними ефектами. На цій траєкторії існуватиме така критична координата r0, коли всі реагуючі атоми зближені й утворюють активований комплекс Х. Активований комплекс з максимальною енергією перебуває в нейстійкому, перехідному стані.
За теорією перехідного стану, швидкість реакції визначається швидкістю проходження через вершину потенціального бар’єра. Припускається, що в реакції
![]() . (4.88)
. (4.88)
Є
певна концентрація активованих комплексів
![]() ,
які перебувають у “квазірівновазі” з
початковими реагентами. Константу
рівноваги можна записати цього процесу
як:
,
які перебувають у “квазірівновазі” з
початковими реагентами. Константу
рівноваги можна записати цього процесу
як:
![]() . (4.89)
. (4.89)
Швидкість
прямої реакції (в напрямку, показаному
стрілкою) визначається середньою
швидкістю
![]() проходження через бар’єр шириною
(рис. 4.34). За розподілом Максвелла
знаходиvj за формулою
проходження через бар’єр шириною
(рис. 4.34). За розподілом Максвелла
знаходиvj за формулою
 (4.90)
(4.90)
де m – маса молекули; Т – температура; k0 – стала Больцмана. Середній час проходження через бар’єр становить:
![]() (4.91)
(4.91)
За квантовою статистикою:
![]() (4.92)
(4.92)
де h – стала Планка. Тоді
![]() (4.93)
(4.93)
У загальному вигляді кінетичне рівняння для реакції (4.88) буде таким:
v = k[A][B], (4.94)
де k – константа швидкості реакції. Разом з тим швидкість реакції – це зміна концентрації за час проходження через потенційний бар’єр
![]() (4.95)
(4.95)
Порівнюючи (4.94) і (4.95) і підставивши (4.93), отримуємо рівняння:
![]() ,
,
з якого знаходимо константу швидкості реакції k
![]() . (4.96)
. (4.96)
Після підстановки константи рівноваги Кр (4.89) маємо
![]() . (4.97)
. (4.97)
Відповідно до рівняння Вант-Гоффа (1.38),
![]()
де G0 – зміна стандартної вільної енергії хімічного перетворення. Тепер можна пов’язати k зі зміною термодинамічних величин, а саме:
![]() . (4.98)
. (4.98)
Оскільки G0 = H0 - TS0, тоді (4.98) буде таким:
![]() (4.99)
(4.99)
Це рівняння Ейрінга.
Знайдемо зв’язок між Н й енергією активації Ea. Для цього прологарифмуємо, а далі знайдемо диференціал по Т від емпіричного рівняння Арреніуса (4.85):
![]() ,
,
![]() (4.100)
(4.100)
і теоретичного рівняння (4.99)
![]() ,
,
![]() . (4.101)
. (4.101)
Порівнюючи (4.100) і (4.101), отримаємо:
Ea = H0 + RT.
Теплова енергія RT реагуючих молекул за Т = 300 К становить 2,5 кДж моль-1, тоді як енергії активації Еа для багатьох хімічних реакцій досягають 100 кДж моль-1. Тому можна вважати, що у формулі Ейрінга (4.99) зміна ентальпії H0 з точністю до RT дорівнює енергії активації Еа.
Термодинамічні величини Н0 і S0 надають інформацію про природу перехідного стану системи й вказують на те, що при активації молекул відбувається значна деформація хімічних зв’язків. Ентропія активації характеризує ймовірність утворення перехідного стану. Коли S0 є великою за значенням й від’ємною, і для утворення перехідного стану реагуючі молекули повинні мати точну просторову орієнтацію й певну конформацію, що для некаталітичних реакцій є мало ймовірно. Фермент значно змінює Н0 і S0, збільшуючи на багато порядків швидкість реакції.
Незважаючи на певні успіхи, теорія абсолютних швидкостей реакцій має низку обмежень. Зокрема, теорія перехідного стану постулює, що енергетичний бар’єр є статичним і його подолання відбувається тільки через енергію співударів атомів. Але білки характеризуються динамічністю своєї структури, й атомні групи постійно зміщуються на 0,05 нм. У щільно упакованій білковій глобулі рухомість атомних груп великого розміру поєднується з колективним зміщенням оточуючих груп. Рухома група співударяється з іншими групами, що створює ефект внутрішнього тертя й сповільнює швидкість переходу через енергетичний бар’єр. За теорією Крамерса (1940), яка базується на моделі руху через бар’єр під дією випадкової сили, постійна швидкості реакції становитиме:
![]() , (4.102)
, (4.102)
де – в’язкість мікросередовища.