

Тема 5. Радикальные и электрофильные реакции органических соединений
Радикальные реакции.
Хлор реагирует с предельными углеводородами только под влиянием света, нагревания или в присутствии катализаторов, причем последовательно замещаются хлором все атомы водорода:
СН4 + С12 СН3С1 + НС1
СН3С1 + С12 СН2С12 + НСl
СН2С12 + С12 CHC13 + HCl
СНС13 + С12 СС14 + НСl
Наиболее легко замещается водород у третичного атома углерода. Соотношение между скоростями замещения (при 300°) водородных атомов при первичном, вторичном и третичном атомах углерода 1 : 3, 25 : 4,3.
Реакция протекает по цепному радикальному механизму.
С12 2С1. зарождение цепи
СН3 – Н + C1. CH3. + HC1 рост, развитие цепи
СН3. + С12 СН3С1 + C1. рост, развитие цепи
2С1. С12 обрыв цепи
СН3. + С1. СН3С1 обрыв цепи
СН3. + СН3. С2Н6 обрыв цепи
Фотохимическое бромирование обычно проходит строго избирательно (селективно) – легче всего замещаются атомы водорода у третичного атома углерода.
Существуют три общих пути генерирования радикальных частиц: расщепление ковалентной связи за счет тепловой энергии (термолиз); расщепление связи при помощи лучистой энергии (фотолиз) и образование радикалов в окислительно-восстановительных процессах.
При сильном нагревании (500°С и выше) тепловой энергии оказывается достаточно для разрыва прочных С—С и С—Н связей. Поэтому большинство процессов при высоких температурах (пиролиз, крекинг) протекает по радикальному механизму.
Облучение видимым или ультрафиолетовым светом часто используется для избирательного (селективного) расщепления относительно слабых связей. Образующиеся при этом радикальные частицы выступают в качестве инициаторов последующих превращений.
Радикальные реакции весьма распространены и в живых системах, так как молекулярный кислород является одним из самых распространенных радикалов и сам способен инициировать радикальные реакции. Известно, что в основном состоянии молекула кислорода представляет собой бирадикал, в котором на каждом атоме кислорода находится по одному неспаренному электрону. Как и другие радикалы, кислород может отрывать Н-атом от углеводородного фрагмента и служить таким образом одним из инициаторов радикальных реакций. В качестве примера можно назвать процессы горения, а также автоокисления, в том числе и в биологических системах.
Когда молекулярный кислород принимает электрон, он превращается в анион-радикал О2-, называемый супероксидом (superoxide). Этот радикал способен участвовать как в полезных, так и в нежелательных физиологических процессах. Например, иммунная система живого организма использует супероксид в своей борьбе с патогенами - чужеродными болезнетворными телами. Вместе с тем супероксид может быть вовлечен в некоторые процессы, вызывающие окислительное повреждение здоровых клеток, что ведет к дегенерации и старению живого организма. Нормальное функционирование системы ферментов предотвращает эти нежелательные процессы.
Один из таких ферментов - супероксид дисмутаза - регулирует уровень супероксида в организме, катализируя его превращение в пероксид водорода и молекулярный кислород. Пероксид водорода, однако, также опасен, поскольку может генерировать гидроксильные радикалы ОН.
Гидроксильный радикал в биологических системах возникает и при неполном восстановлении молекулы кислорода
О2 + 3е- + 3Н+ НО + Н2О
Пероксидные радикалы возникают при взаимодействии молекулы кислорода с ионами тяжелых металлов, например ионами железа(II)
Fe2+ + О2 + Н+ Fe3+ + Н–О–О гидропероксильный радикал
Пероксильные и гидроксильные радикалы действуют избирательно (селективно), атакуя в ненасыщенных высших кислотах связи С—Н метиленовых групп, соседних с двойной связью. При этом образуются наиболее стабильные в данном случае радикалы аллильного типа.
С6Н13СН(ОН)–СН2–СН=СН–СН2–(СН2)6СООН
С6Н13СН(ОН)– СН–СН=СН–СН2–(СН2)6СООН + С6Н13СН(ОН)–СН2–СН=СН–СН–(СН2)6СООН
Свободные радикалы аллильного типа могут вступать в различные реакции, в частности с молекулами кислорода и воды, с образованием гидропероксидов.
Аллильный радикал + О2 С6Н13СН(ОН)– (ОО)СН–СН=СН–СН2–(СН2)6СООН
Пероксильный радикал + Н2О С6Н13СН(ОН)– (НОО)СН–СН=СН–СН2–(СН2)6СООН + ОН
Гидропероксиды неустойчивы. Они подвергаются дальнейшим превращениям. При этом сначала образуются альдегиды, которые легко окисляются в кислоты. Таким образом, наличие свободных радикалов в организме вызывает цепь реакций, изменяющих структуру и, следовательно, функции кислоты.
Ненасыщенные высшие жирные кислоты — структурные компоненты клеточных мембран. Свободные радикалы являются мощным фактором, повреждающим клеточные мембраны.
Фермент каталаза, также присутствующий в живом организме, предотвращает образование ОН-радикалов.
Эти процессы, протекающие в живой клетке, показывают, что в соответствующих ситуациях важным является не только инициирование, но и ингибирование радикальных реакций.
В живых организмах ингибиторами окислительных реакций также могут выступать природные антиоксиданты - гидрокси- и полигидроксисоединения.
Например, -токоферол (витамин Е) действует как ловушка
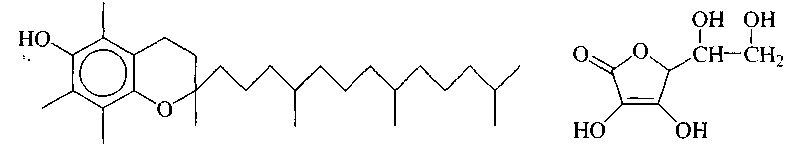
витамин Е (-токоферол) витамин С
радикалов и ингибирует нежелательные радикальные процессы в организме, способные вызвать повреждение клеток.
Витамин С также является антиоксидантом, эффективно работающим в живых системах. Тем не менее и этим антиоксидантом нельзя злоупотреблять: не следует принимать более 500 мг витамина С однократно .
Реакции электрофильного присоединения
Ненасыщенные углеводороды — алкены, циклоалкены, алкадиены и алкины — проявляют способность к реакциям присоединения, так как содержат двойные или тройные связи. Более важной in vivo является двойная связь.
За счет электронов -связи в молекулах алкенов имеется область повышенной электронной плотности. Поэтому они представляют собой нуклеофилы и, следовательно, склонны подвергаться атаке электрофильным реагентом.
Механизм реакции. Присоединение к алкенам галогеноводородов и родственных соединений протекает по гетеролитическому электрофильному механизму ае. Электрофильной частицей в данном процессе часто служит простейший электрофил — протон Н+. В реакции выделяют две основные стадии: 1) электрофильная атака протоном алкена с образованием карбокатиона — медленная стадия, определяющая скорость процесса в целом; 2) атака образовавшегося карбокатиона нуклеофилом Х—, приводящая к конечному продукту (быстрая стадия).
Присоединение галогеноводородов и воды (гидратация) происходит по правилу Марковникова: атомы водорода присоединяются преимущественно к наиболее гидрогенизированному атому углерода (наиболее богатому водородом), а атом галогена или гидроксигруппа присоединяются к наименее гидрогенизированному.
Н3С–СН = СН2 + НCl Н3С–СНCl–СН3
Образуется преимущественно 2-хлорпропан, а не 1-хлорпропан.
Но такая формулировка правила Марковникова не объясняет направление реакций гипогалогенирования (присоединения НОГал) и реакций присоединения к алкенам, в состав которых входят группы, обладающие отрицательным мезомерным или индукционным эффектами.
Существует и другая более общая формулировка правила Марковникова: присоединение несимметричного реагента к несимметричному алкену по ионному механизму протекает в направлении образования более устойчивого катиона. Так, в результате присоединения катиона водорода к двойной связи пропена могут возникнуть два карбокатиона:
СН3СН2+СН2 (I) и СН3+СНСН3 (II)
Ни в том, ни в другом катионе положительный заряд на выделенном атоме углерода не равен, а много меньше +1 из-за смещения к нему электронной плотности от соседних атомов. Но в первичном катионе (I) донором этой плотности является один, а во вторичном (II) – два соседних атома углерода. Кроме того, в первичном катионе (I) метильная группа находится дальше, ее индукционный эффект по цепи затухает. Это значит, что распределение заряда во вторичном карбокатионе (II) более равномерное; такая частица оказывается более устойчивой, реакция идет по пути, направляемому ее образованием.
Рассмотрим присоединение галогеноводорода к непредельным карбоновым кислотам:
СН2 = СН – СООН + НCl СН2Cl – СН2 – СООН
В этом случае водород присоединяется к менее гидрогенизированному атому углерода. Значит ли это что присоединение идет против правила Марковникова? Оказывается, нет. В результате присоединения иона водорода к двойной связи кислоты могут возникнуть два карбокатиона:
+СН2 – СН2 СООН (I) и СН3 +СН СООН (II)
В первичном катионе (I) нет донорной метильной группы, зато дестабилизирующее действие акцепторной карбоксильной группы ощущается заметно меньше, чем во вторичном катионе (II). Отрицательный мезомерный и индукционный эффекты в карбоксильной группе увеличивают положительный заряд во вторичном катионе (II) в большей степени, чем в первичном (I). Это значит, что распределение заряда в первичном карбокатионе (I) более равномерное; такая частица оказывается более устойчивой, реакция идет по пути, направляемому ее образованием. Присоединение фактически следует правилу Марковникова в его обобщенной форме.
В случае реакции гипохлорирования (присоединения HOCl) в растворе присутствуют ионы ОН- и Cl+, а протона нет совсем и применима только обобщенная формулировка правила Марковникова. Тогда в результате присоединения катиона хлора к двойной связи пропена могут возникнуть два карбокатиона, из которых более устойчивым будет являться вторичный (II):
СН3СНCl +СН2 (I) и СН3+СН СН2Cl (II)
Тогда реакцию можно записать следующим образом:
Н3С–СН=СН2 + НОCl Н3С–СН(ОН)–СН2Cl
Присоединение против правила Марковникова идет в том случае, если реакцию проводят в присутствии перекисей (Н2О2 или ROOR). Тогда реакция протекает по другому механизму (свободнорадикальное присоединение):
Н 3С–СН=СН2
+ НBr
ROOR
Н3С–СН2–СН2Br
3С–СН=СН2
+ НBr
ROOR
Н3С–СН2–СН2Br
Для сопряженных диенов характерна способность образовывать в реакциях присоединения наряду с обычными 1,2-аддуктами продукты 1,4-присоединения. Соотношение между ними в значительной степени зависит от условий эксперимента.
Реакции электрофильного замещения. Для ароматических соединений бензольного ряда, конденсированных и гетероциклических ароматических соединений характерны реакции, не приводящие к нарушению ароматической системы, т. е. реакции замещения. Они не склонны вступать в реакции присоединения или окисления, ведущие к нарушению ароматичности.
Для соединений ароматического характера, имеющих замкнутую -электронную систему, наиболее характерны реакции с электрофильными агентами.
С помощью кинетических методов показано, что большинство реакций электрофильного замещения в ароматическом ряду протекает по двухстадийному механизму. На первой, медленной, стадии происходит нарушение ароматической системы и переход атакуемого атома углерода ядра в состояние sp3-гибридизации. Вторая, быстрая, стадия сопровождается восстановлением ароматической структуры и вследствие выигрыша энергии протекает легко и быстро.

Следует заметить, что протекание первой стадии обычно осуществляется с промежуточным образованием так называемого -комплекса. -Комплексы представляют собой координационные соединения, в которых донором являются ароматические соединения, имеющие легко поляризуемые -электроны, а акцепторами – галогены, сильные минеральные кислоты и тому подобные соединения, имеющие по разным причинам сродство к электронам.
-Комплексы не являются истинными химическими соединениями, в которых электрофильная частица связывается ковалентной связью с конкретным атомом субстрата. -Комплексы, образуемые бензолом, очевидно, построены объемно, так как наибольшая -электронная плотность ароматических соединений расположена по обе стороны бензольного кольца.
Первой ступенью взаимодействия ароматических соединений с электрофильным агентом Е+ при реакциях электрофильного замещения является образование - комплекса, который медленно, лимитируя скорость всей реакции, перегруппировывается в карбкатион (- комплекс).
В отличие от -комплексов, -комплексы представляют собой истинно химические соединения, в которых электрофильный агент Е+ образует ковалентную связь за счет двух -электронов одной из связей бензольного кольца. При образовании -комплекса электрофильный агент внедряется в ароматическую молекулу более «глубоко», чем в -комплексе. В -комплексе один из атомов углерода бензольного кольца переходит в состояние sp 3-гибридизации, в котором все четыре связи направлены под углом 109°. При этом нарушается симметрия бензольного кольца, а группа Е+ и атом водорода оказываются в положении, перпендикулярном плоскости кольца.
Реакции электрофильного замещения завершаются отщеплением от -комплекса протона и восстановлением ароматической системы. Как правило, этот процесс происходит при участии обладающего основными свойствами аниона А–, присутствующего в реакционной среде, и сопровождается выделением энергии.
К реакциям электрофильного замещения относятся реакции галогенирования, нитрования, сульфирования, алкилирования, ацилирования:
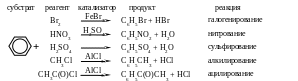
Заместители в кольце делят на ориентанты I рода и II рода. Ориентанты I рода: алкильные радикалы, группы –ОН, –OR, –NH2, –NHR, –NR2, –SH, атомы галогенов направляют второй заместитель в орто- и пара-положения. Все заместители I рода, кроме галогенов, обладают электронодонорными свойствами по отношению к бензольному кольцу и ускоряют реакции электрофильного замещения по сравнению с незамещённым бензолом.
Ориентанты II рода: карбонильные и сложноэфирные группы, группы –СООН, –SO3H, –NO2, –CN направляют второй заместитель в мета-положение. Все заместители II рода обладают электроноакцепторными свойствами по отношению к бензольному кольцу и замедляют реакции электрофильного замещения по сравнению с незамещённым бензолом.
Контрольные задания
1. Укажите, какая из стадий хлорирования метана на свету показывает обрыв цепи:
а) СН4 + С1 СН3 + НС1; б) СН3 + С12 СН3С1 + Сl;
в) СН3С1+С1 СН2С1+НС1; г) СН3 + СН3 CH3 – CH3.
К какому типу относится эта реакция?
2. Напишите уравнения бромирования, сульфирования, и нитрования бутана.
3. По какому механизму идёт реакция нитрования алканов:
а) радикального замещения; б) нуклеофильного присоединения; в) радикального присоединения;
в) электрофильного присоединения; г) электрофильного замещения?
4. Напишите уравнения реакции присоединения бромоводорода к 1-бутену в присутствии перекиси и без нее. В виде каких стереоизомеров может существовать исходное соединение? Образуют ли стереоизомеры продукты реакции?
5. Какое соединение образуется при гидратации 1-бутена:
а) 1-бутанол; б) 1-бутин; в) 2-бутанол; г) бутан; д) 2-бутен.
6. Взаимодействие воды с алкенами протекает по правилу Марковникова или против него?
7. Какое вещество преимущественно образуется при присоединении брома к 1,3-бутадиену при температуре минус 80 С? А при температуре + 40 С? Приведите уравнения реакций.
8. Приведите схемы алкилирования хлорбензола, фенола, нитробензола. Расположите эти соединения в ряд по увеличению скорости реакции.
9. Следующие соединения расположите в ряд по увеличению реакционной способности при бромировании их в бензольное кольцо: а) бензол; б) фенол; в) бензальдегид; г) этилбензол. Дайте объяснения.
10. Определите положения, по которым преимущественно будет проходить нитрование м- нитротолуола и п- нитротолуола. Отметьте тип ориентации заместителей (совпадающая или несовпадающая).