

17Билет.
Для
объяснения отличия валентных углов в
молекулах H2O (104,5°) и NH3 (107,3°) от 90° следует
принять во внимание, что устойчивому
состоянию молекулы отвечает ее
геометрическая структура с наименьшей
потенциальной энергией. Поэтому при
образовании молекулы форма и взаимное
расположение атомных электронных
облаков * изменяется по сравнению с их
формой и расположением в свободных
атомах. В результате достигается более
полное перекрывание орбиталей * при
образовании химической связи. Такая
деформация электронных облаков требует
затраты энергии, но более полное
перекрывание приводит к образованию
более прочной связи, и в целом получается
выигрыш в энергии. Этим и объясняется
возникновение гибридных орбиталей.
Форма гибридной орбитали может быть
определена математически путем сложения
волновых функций * исходных орбиталей:
![]()
В результате сложения волновых функций s- и p-орбиталей с учетом их знаков оказывается, что плотность электронного облака (величина |y|2) по одну сторону от ядра повышена, а по другую – понижена.
В целом процесс гибридизации включает следующие этапы: возбуждение атома *, гибридизация орбиталей возбужденного атома, образование связей с другими атомами. Затраты энергии на первые два этапа компенсируются выигрышем энергии при образовании более прочных связей с гибридными орбиталями. Тип гибридизации определяется типом и количеством участвующих в ней орбиталей.
Ниже рассмотрены примеры различных видов гибридизации s- и p-орбиталей..Гибридизация одной s- и одной p-орбитали (sp-гибридизация) происходит, например, при образовании галогенидов бериллия, цинка, кадмия и ртути. Атомы этих элементов в нормальном состоянии имеют во внешнем слое два спаренных s-электрона. В результате возбуждения один из s-электронов переходит в p-состояние – появляется два неспаренных электрона, один из которых s-, а другой p-электрон. При образовании химической связи * эти две различные орбитали преобразуются в две одинаковые гибридные орбитали (sp-орбитали), направленные под углом 180° друг к другу, – две связи имеют противоположное направление
![]()
Экспериментальное определение структуры молекул BeГ2, ZnГ2, CdГ2, HgГ2 (Г–галоген) показало, что эти молекулы являются линейными, и обе связи металла с атомами галогена имеют одинаковую длину.
Г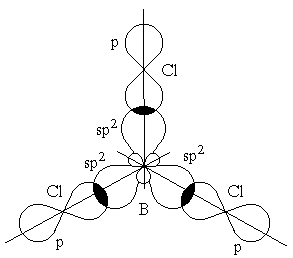 ибридизация
одной s- и двух p-орбиталей (sp2-гибридизация)
имеет место, например, при образовании
соединений бора. Возбужденный атом
бора обладает тремя неспаренными
электронами – одним s-электроном и
двумя p-электронами. Из трех орбиталей
образуются три эквивалентные sp2-гибридные
орбитали, расположенные в одной плоскости
под углом 120° друг к другу (рисунок 3.6).
Действительно, как показывают
экспериментальные исследования,
молекулы таких соединений бора, как
BГ3 (Г-галоген), B(CH3)3 – триметилбор, B(OH)3
– борная кислота, имеют плоское строение.
При этом три связи бора в указанных
молекулах имеют одинаковую длину и
расположены под углом 120°.
ибридизация
одной s- и двух p-орбиталей (sp2-гибридизация)
имеет место, например, при образовании
соединений бора. Возбужденный атом
бора обладает тремя неспаренными
электронами – одним s-электроном и
двумя p-электронами. Из трех орбиталей
образуются три эквивалентные sp2-гибридные
орбитали, расположенные в одной плоскости
под углом 120° друг к другу (рисунок 3.6).
Действительно, как показывают
экспериментальные исследования,
молекулы таких соединений бора, как
BГ3 (Г-галоген), B(CH3)3 – триметилбор, B(OH)3
– борная кислота, имеют плоское строение.
При этом три связи бора в указанных
молекулах имеют одинаковую длину и
расположены под углом 120°.
Гибридизация одной s- и трех p-орбиталей (sp3-гибридизация) характерна, например, для углерода и его аналогов – кремния и германия. В этом случае четыре гибридные sp3-орбитали расположены под углом 109°28¢ друг к другу; они направлены к вершинам тетраэдра (в молекулах CH4, CCl4, SiH4, GeBr4 и др.). Валентные углы в молекулах H2O (104,5°) и NH3 (107,3°) не точно соответствуют взаимному расположению “чистых” p-орбиталей (90°). Это обусловлено некоторым вкладом s-электронов в образование химической связи. Такой вклад есть не что иное, как гибридизация. Валентные электроны в этих молекулах занимают четыре орбитали, которые близки к sp3-гибридным. Незначительное отличие валентных углов от тетраэдрических 109°28¢ объясняется тем, что гибридизация в данном случае является неполной.
Во многих молекулах центральный атом не подвергается гибридизации. Так, валентные углы в молекулах H2S, PH3 и др. близки к 90°, т.е. образование связей происходит с участием “чистых” p-орбиталей, расположенных под прямым углом друг к другу.
Водородная связь
Часто водородную связь рассматривают как электростатическое взаимодействие, усиленное небольшим размером водорода, которое разрешает близость взаимодействующих диполей. Тогда об этом говорят как о разновидности донорно-акцепторной связи, невалентном взаимодействии между атомом водорода H, ковалентно связанным с атомом A группы A-H молекулы RA-H и электроотрицательным атомом B другой молекулы (или функциональной группы той же молекулы) BR'. Результатом таких взаимодействий являются комплексы RA-H•••BR' различной степени стабильности, в которых атом водорода выступает в роли «моста», связывающего фрагменты RA и BR'.
Особенностями водородной связи, по которым её выделяют в отдельный вид, является её не очень высокая прочность[2], её распространенность и важность, особенно в органических соединениях[3], а также некоторые побочные эффекты, связанные с малыми размерами и отсутствием дополнительных электронов у водорода.
В настоящее время в рамках теории молекулярных орбиталей водородная связь рассматривается как частный случай ковалентной с делокализацией электронной плотности по цепи атомов и образованием трёхцентровых четырёхэлектронных связей (например, -H•••[F-H•••F]-).
Водородная связь в значительной мере определяет свойства и таких биологически важных веществ, как белки и нуклеиновые кислоты. В частности, элементы вторичной структуры (например, α-спирали, β-складки) и третичной структуры в молекулах белков, РНК и ДНК стабилизированы водородными связями. В этих макромолекулах, водородные связи сцепляют части той же самой макромолекулы, заставляя её сворачиваться в определенную форму. Например, двойная спиральная структура ДНК, определяется в значительной степени наличием водородных связей, сцепляющих пары нуклеотидов, которые связывают одну комплементарную нить с другой. Много полимеров усилены водородными связями в их главных цепях. Среди синтетических полимеров самый известный пример - нейлон, где водородные связи играют главную роль в кристаллизации материала. Водородные связи также важны в структуре полученных искусственно полимеров (например, целлюлозы) и в многих различных формах в природе, таких как древесина, хлопок и лён.
18-билет
Комплексные соединения или координационные соединения— частицы (нейтральные молекулы или ионы), которые образуются в результате присоединения к данному иону (или атому), называемому комплексообразователем, нейтральных молекул или других ионов, называемых лигандами. Теория комплексных соединений (координационная теория) была предложена в 1893 г. А. Вернером. Комплексные соединения мало диссоциируют в растворе (в отличие от двойных солей). Комплексные соединения могут содержать комплексный малодиссоциирующий анион ([Fe(CN)6]3−), комплексный катион ([Ag(NH3)2]+) либо вообще не диссоциировать на ионы (соединения типа неэлектролитов, например карбонилы металлов). Комплексные соединения разнообразны и многочисленны.
омплексное соединение – химическое вещество, в состав которого входят комплексные частицы. В настоящее время строгого определения понятия " комплексная частица" нет. Обычно используется следующее определение. Комплексная частица – сложная частица, способная к самостоятельному существованию в кристалле или растворе, образованная из других, более простых частиц, также способных к самостоятельному существованию. Иногда комплексными частицами называют сложные химические частицы, все или часть связей в которых образованы по донорно-акцепторному механизму.
Комплексообразователь – центральный атом комплексной частицы. Обычно комплексообразователь – атом элемента, образующего металл, но это может быть и атом кислорода, азота, серы, йода и других элементов, образующих неметаллы. Комплексообразователь обычно положительно заряжен и в таком случае именуется в современной научной литературе металлоцентром; заряд комплексообразователя может быть также отрицательным или равным нулю.
Лиганды – атомы или изолированные группы атомов, располагающиеся вокруг комплексообразователя. Лигандами могут быть частицы, до образования комплексного соединения представлявшие собой молекулы (H2O, CO, NH3 и др.), анионы (OH−, Cl−, PO43− и др.), а также катион водорода H+.
Внутренняя сфера комплексного соединения – центральный атом со связанными с ним лигандами, то есть, собственно, комплексная частица.
Внешняя сфера комплексного соединения – остальные частицы, связанные с комплексной частицей ионной или межмолекулярными связями, включая водородные.
Дентатность лиганда определяется числом координационных мест, занимаемых лигандом в координационной сфере комплексообразователя. Различают монодентатные (унидентатные) лиганды, связанные с центральным атомом через один из своих атомов, то есть одной ковалентной связью), бидентатные (связанные с центральным атомом через два своих атома, то есть, двумя связями), три- , тетрадентатные и т.д.
Координационный полиэдр – воображаемый молекулярный многогранник, в центре которого расположен атом
комплексообразователь, а в вершинах – частицы лигандов, непосредственно связанные с центральным атомом.
Координационное число (КЧ) – число - связей, образуемых центральным атомом с лигандами. Для комплексных соединений с монодентантными лигандами КЧ равно числу лигандов, а в случае полидентантных лигандов - числу таких лигандов, умноженному на дентатность.
Диссоциация
ионов [Ag(NH3)2]+, согласно приведенному
выше уравнению, как и диссоциация
всякого электролита, подчиняется закону
действия масс и может быть охарактеризована
соответствующей константой равновесия,
называемой константой нестойкости
комплексного иона:
![]() Константы
нестойкости для различных комплексных
ионов весьма различны и могут служить
мерой устойчивости комплекса.Константы
нестойкости, в выражения которых входят
концентрации ионов и молекул, называются
«концентрационными». Более строгими
и не зависящими от концентраций и ионной
силы раствора являются константы
нестойкости, содержащие вместо
концентраций активности ионов и молекул.
В разбавленных растворах эти два
различных выражения констант нестойкости
совпадают друг с другом.
Константы
нестойкости для различных комплексных
ионов весьма различны и могут служить
мерой устойчивости комплекса.Константы
нестойкости, в выражения которых входят
концентрации ионов и молекул, называются
«концентрационными». Более строгими
и не зависящими от концентраций и ионной
силы раствора являются константы
нестойкости, содержащие вместо
концентраций активности ионов и молекул.
В разбавленных растворах эти два
различных выражения констант нестойкости
совпадают друг с другом.
Из приведенной формулы видно:
Чем меньше концентрация продуктов распада, т.е. чем устойчивее комплекс, тем меньше его константа нестойкости. Наиболее устойчивые в растворах комплексные частицы имеют наименьшие константы нестойкости.
19-билет
1).Химическая связь в комплексных соединениях и особенности их строения. В образовании комплексных соединений важную роль играют донорно - акцепторные взаимодействия лиганда и центрального атома. Донором электронной пары, как правило, является лиганд. Акцептором – центральный атом, который имеет свободные орбитали. Связь эта прочна и не разрывается при растворении комплекса (неионогенна) и ее называют координационной.
Наряду с s-связями образуются p-связи по донорно-акцепторному механизму. При этом донором служит ион металла отдающий свои спаренные d-электроны лиганду, имеющему энергетически выгодные вакантные орбитами. Такие связи называют дативными. Они образуются: а) за счет перекрывания вакантных р-орбиталей металла с d-орбиталью металла на которой находятся электроны не вступившие в s-связь (dp-pp взаимодействие); б) при перекрывании вакантных d-орбиталей лиганда с заполненными d-орбиталями металла .Мерой ее прочности является степень перекрывания орбиталей лиганда и центрального атома. Направленность связей центрального атома определяет геометрию комплекса (положение лигандов в пространстве). Для объяснения направленности связей используются представления о гибридизации атомных орбиталей центрального атома. Орбитали комплексообразователя, участвующие в образовании связи, подвергаются гибридизации. Гибридные орбитали центрального атома являются результатом смешения неравноценных атомных орбиталей, в результате форма и энергия орбиталей взаимно изменяется и образуются орбитали новой, но уже одинаковой формы и энергии. Число гибридных всегда равно числу исходных. Гибридные облака располагаются в атоме как можно дальше друг от друга. Различают следующие типы гибридизации атомных орбиталей комплексообразователя. Центральный атом образует sp гибридные обитали, которые располагаются под углом 180 град., что соответствует его кч=2. Геометрия таких молекул линейная. sp3 гибридизация соответствует кч=4. Геометрия комплекса тетраэдрическая. sp3d2 гибридизация определяет кч центрального атома равное 6 и геометрию комплекса октаэдрическую. Применение комплексонов и комплексонатов в медицине. Вещества, устраняющие последствия воздействия ядов на биологические структуры и инактивирующие яды, посредством химических реакций, называют антидотами. В настоящее время применяют унитиол. Этот препарат эффективно выводит из организма мышьяк, ртуть, хром и висмут. Наиболее широко используют при отравлении цинком, кадмием, свинцом и ртутью комплексоны и комплексонаты. Применение их основано на образовании более прочных комплексов с ионами металлов, чем комплексы этих же ионов с серосодержащими группами белков, аминокислот и углеводов. Для выведения свинца используют препараты на основе ЭДТА. Введение в организм в больших дозах препаратов опасно, так как они связывают ионы кальция, что приводит к нарушению многих функций. Поэтому применяют тетацин, CaNa2 ЭДТА, который используют для выведения свинца, кадмия, ртути, итрия, церия и др. редкоземельных металлов и кобальта.
Ионы токсиканты вытесняют кальций из тетацина в связи с образованием более прочных связей с кислородом и ЭДТА.
Билет 20
1.
Электро́дный
потенциа́л —
разность электрических
потенциалов между электродом и
находящимся с ним в контакте электролитом(чаще
всего между металлом и раствором электролита).
Возникновение электродного потенциала
обусловлено переносом заряженных
частиц через границу раздела фаз,
специфической адсорбцией ионов,
а при наличии полярных молекул
— ориентационной адсорбцией их. Величина
электродного потенциала в неравновесном
состоянии зависит как от природы и
состава контактирующих фаз, так и от
кинетических закономерностей электродных
реакций на границе раздела фаз.
Зависимость
электродного потенциала от
термодинамических активностей участников
электрохимических реакции выражается Нернста
уравнением:![]() где
vi -
стехиометрический коэфицент участника
реакции, причем для исходных веществ
это отрицательная величина, а для
продуктов реакции положительная. Если
через электрод протекает
электрический ток, электродный потенциал
отклоняется от равновесного значения
из-за конечной скорости процессов,
происходящих непосредственно на
границе электрод -электролит.
где
vi -
стехиометрический коэфицент участника
реакции, причем для исходных веществ
это отрицательная величина, а для
продуктов реакции положительная. Если
через электрод протекает
электрический ток, электродный потенциал
отклоняется от равновесного значения
из-за конечной скорости процессов,
происходящих непосредственно на
границе электрод -электролит.
Окислительно-восстановительный потенциал — мера способности химического вещества присоединять электроны (восстанавливаться). Окислительно-восстановительный потенциал выражают в милливольтах (мВ). Окислительно-восстановительный потенциал определяют как электрический потенциал, устанавливающийся при погружении платины или золота(инертный электрод) в окислительно-восстановительную среду, то есть в раствор, содержащий как восстановленное соединение(Ared), так и окисленное соединение (Aox). Редокс-электроды состоят из электрохимически инертного проводника (платины, графита и т. д.), погруженного в раствор, в котором находятся окисленная и восстановленная формы потенциалопределяющего вещества. Примерами таких электродов могут служить редокс-электроды с ионами в различных степенях окисления: (Pt)Sn4+, Sn2+, (Pt)Fe3+, Fe2+
![]()
![]()
Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала индикаторного электрода. Это наблюдается, конечно, лишь тогда когда хотя бы один из участников реакции титрования является участником электродного процесса. Так, например, титрование по методу кислотно-основного взаимодействия может быть выполнено со стеклянным электродом. Определение хлорида - с хлорсеребряным и т.д. Виды потенциометрического титрования. Кислотно-основное титрование. В кислотно-основном титровании в качестве индикаторного обычно используют стеклянный электрод входящий в комплект серийно выпускаемых промышленностью pH-метров. Комплексонометрическое титрование. Потенциометрическое титрование катионов комплексоном III (ЭДТА) можно проводить с использованием в качестве индикаторного электрода соответствующего металла: титрование солей меди с медным электродом, солей цинка с цинковым и т.д. или подходящего ионоселективного электрода. Титрование по методу осаждения. Индикаторными электродами в методах потенциометрического титрования, использующих реакции осаждения, служат металлические или мембранные электроды, чувствительные к определяемому иону или иону-осадителю.
Роль ОВР в жизнедеятельности организма. Пероксид водорода применяют в пищевой отрасли промышленности для отбеливания шоколада, рубцов и оболочек в производстве сосисок.
2. Mg+12. 1s 22s 22p 63s 2. )1s2)2s2 2p6)3s2. 10Ne[ 3s2 ] (степень окисления +2)
 2
8 2
2
8 2
n=1 n=2 n=3
L=0 L=0,1 L=0,1,2
me=1 me=1,3 me=1,3,5
ms= -1/2,+1/2 ms= -1/2,+1/2 ms= -1/2,+1/2
Физические и химические свойства: металлический магний обладает гексагональной кристаллической решеткой. Поверхность магния покрыта плотной пленкой оксида MgO. При нагревании металла до температуры выше примерно 600°C он загорается на воздухе. Оксид магния MgO представляет собой белый рыхлый порошок, не реагирующий с водой. Раньше его называли жженой магнезией или просто магнезией.
MgO + 2HNO3 = Mg(NO3)2 + H2O.
2NaOH + MgSO4 = Mg(OH)2 + Na2SO4.
Mg2C3 + 4Н2О = 2Mg(OH)2 + С3Н4.
3Mg + N2= Mg3N2.
Mg3N2 + 6Н2О = 3Mg(ОН)2 + 2NН3.
2Mg(HCO3)2 = (MgOH)2CO3 + 3CO2 + Н2О.
Нахождение в природе: магний — один из десяти наиболее распространенных элементов земной коры. Из-за высокой химической активности в свободном виде магний не встречается, а входит в состав множества минералов — силикатов, алюмосиликатов, карбонатов, сульфатов и др.
Применение: основная часть добываемого магния используется для получения различных легких магниевых сплавов. В состав этих сплавов, кроме магния, входят алюминий, цинк, цирконий. Эти сплавы достаточно прочны и находят применение в самолетостроении, приборостроении и д.р.
Биологическая роль: магний — биогенный элемент, постоянно присутствующий в тканях всех организмов. Он входит в состав молекулы зеленого пигмента растений — хлорофилла, участвует в минеральном обмене, активирует ферментные процессы в организме, повышает засухоустойчивость растений. В организм животных и человека магний поступает с пищей. В медицине применяют препараты магния — его сульфат, карбонат, жженую магнезию.
3. Восстанавливающие дисахариды. В дисахаридах один из моносахаридных остатков участвует в образовании гликозидной связи за счет гидроксильной группы чаще всего при С-4 или С-6, реже при С-З. В дисахариде имеется свободная полуацетальная гидроксильная группа, вследствие чего сохраняется способность к раскрытию цикла. Восстановительные свойства таких дисахаридов обусловлены возможностью осуществления цикло-оксо-таутомерии. К восстанавливающим дисахаридам относятся мальтоза, целлобиоза, лактоза. При гидролизе расщепляются на составляющие их моносахариды за счёт разрыва гликозидны связей между ними.
4.
Спирты́ — органические
соединения,
содержащие одну или более гидроксильных
групп непосредственно
связанных с насыщенным атомом углерода.
Спирты можно рассматривать как
производные воды
(H−O−H),
в которых один атом водорода замещен
на органическую функциональную
группу: R−O−H.
Этерификация —
реакция образования сложных
эфиров при взаимодействии
кислот и спиртов:
RCOOH
+ R’OH ⇔
RCOOR' + Н2О.
Кислотные
свойства спиртов.
Как
слабые кислоты,
спирты способны диссоциировать по
связи O−H с образованием алкоксид-иона![]() .
Спирты
могут образовывать с сильными минеральными
кислотами соли алкоксония, а также
давая донорно-акцепторные комплексы
с кислотами
.
Спирты
могут образовывать с сильными минеральными
кислотами соли алкоксония, а также
давая донорно-акцепторные комплексы
с кислотами
![]() Спирты довольно слабые основания и их
относительная основность, в отличие
от кислотности, сохраняется как в
растворе, так и газовой фаза. CH3OH
< CH3CH2OH
< CH3CH2CH2OH
< (CH3)2CHOH
< (CH3)3COH.
Межмолекулярная
и внутримолекулярная дегидратация
спиртов.
При осторожном нагревании в
присутствии серной
кислоты происходит
межмолекулярная дегидратация спиртов
с образованием простых
эфиров.
Если
в реакцию с кислотой вступают двухатомные
спирты, будет протекать реакция
внутримолекулярной дегидратации с
образованием гетероциклических
соединений:
Спирты довольно слабые основания и их
относительная основность, в отличие
от кислотности, сохраняется как в
растворе, так и газовой фаза. CH3OH
< CH3CH2OH
< CH3CH2CH2OH
< (CH3)2CHOH
< (CH3)3COH.
Межмолекулярная
и внутримолекулярная дегидратация
спиртов.
При осторожном нагревании в
присутствии серной
кислоты происходит
межмолекулярная дегидратация спиртов
с образованием простых
эфиров.
Если
в реакцию с кислотой вступают двухатомные
спирты, будет протекать реакция
внутримолекулярной дегидратации с
образованием гетероциклических
соединений:
Билет 21
Поверхностная
свободная энергия,
энергия, сосредоточенная на границе
раздела фаз, избыточная по сравнению
с энергией в объеме. При увеличении
пов-сти раздела фаз уд. полная поверхностная
энергия (на единицу пов-сти) e характеризует
увеличение энергии системы. Она равна
сумме механической работы s образования
единицы площади пов-сти и поглощаемой
при этом теплоты q. B обратимом изотермич.
процессе ![]() ,
где Т-абс. т-ра, —
,
где Т-абс. т-ра, —![]() -уд.
поверхностная энтропия.
Поверхностный слой жидкости обладает
определенным запасом поверхностной
энергии. Эта энергия затрачивается на
работу, направленную на образование
поверхности раздела фаз. При постоянном
делении она соотвествует свободной
энергии Гиббса - поверхностная энергия
Гиббса. Поверхностное натяжение (σ)
– это величина, измеряемая свободной
энергией Гиббса, приходящейся на единицу
площади поверхностного слоя (дж/м2).
Оно численно равно работе, которую
необходимо совершить для образования
единицы поверхности раздела фаз при
постоянной температуре. ПН-это сила,
стремящаяся сократить свободную
поверхность тела до наименьших возможных
размеров при данном объеме (H/м).
Поверхностная энергия Es=
σ*S.
σ-поверхностное натяжение. S-
площадь межфазной поверхности.
Поверхностно-активные вещества - это
вещества, при растворении которых
понижается поверхностное натяжение
на границе раздела фаз(спирты,
алифатические кислоты, сложные эфиры):
σр-ра <
σ0
;
Δσ
<
0;
g
>
0; Г > 0. Поверхностно-неактивные вещества
– это вещества, растворение которых
практически не изменяет поверхностное
натяжение на границе раздела фаз
(дисахариды, глюкоза): σр-ра
= σ0;
Δσ
=
0;
g
≈
0; Г = 0. Ориентация
молекул ПАВ
определяет возможность создания и
стабилизации как обычных буровых
растворов, так и утяжеленных с твердой
фазой из частиц неглинистых пород.
Правило распределения полярностей
разъясняет порядок ориентации
молекул ПАВ на
границе раздела твердое тело - жидкость.
При этом полярная часть молекулы ПАВ
будет обращена к полярной фазе, а
неполярная часть - к неполярной. С
увеличением расстояния от твердой
поверхности ориентация
молекул поверхностно-активного
вещества ПАВ
нарушается, а затем пропадает. Толщина
граничного слоя зависит от строения
молекул и внешних условий. Максимальное
значение краевого угла отвечает полному
насыщению адсорбционного слоя, когда
достигается наиболее
полная предельная ориентация молекул
ПАВ гидрофобными
концами наружу.
-уд.
поверхностная энтропия.
Поверхностный слой жидкости обладает
определенным запасом поверхностной
энергии. Эта энергия затрачивается на
работу, направленную на образование
поверхности раздела фаз. При постоянном
делении она соотвествует свободной
энергии Гиббса - поверхностная энергия
Гиббса. Поверхностное натяжение (σ)
– это величина, измеряемая свободной
энергией Гиббса, приходящейся на единицу
площади поверхностного слоя (дж/м2).
Оно численно равно работе, которую
необходимо совершить для образования
единицы поверхности раздела фаз при
постоянной температуре. ПН-это сила,
стремящаяся сократить свободную
поверхность тела до наименьших возможных
размеров при данном объеме (H/м).
Поверхностная энергия Es=
σ*S.
σ-поверхностное натяжение. S-
площадь межфазной поверхности.
Поверхностно-активные вещества - это
вещества, при растворении которых
понижается поверхностное натяжение
на границе раздела фаз(спирты,
алифатические кислоты, сложные эфиры):
σр-ра <
σ0
;
Δσ
<
0;
g
>
0; Г > 0. Поверхностно-неактивные вещества
– это вещества, растворение которых
практически не изменяет поверхностное
натяжение на границе раздела фаз
(дисахариды, глюкоза): σр-ра
= σ0;
Δσ
=
0;
g
≈
0; Г = 0. Ориентация
молекул ПАВ
определяет возможность создания и
стабилизации как обычных буровых
растворов, так и утяжеленных с твердой
фазой из частиц неглинистых пород.
Правило распределения полярностей
разъясняет порядок ориентации
молекул ПАВ на
границе раздела твердое тело - жидкость.
При этом полярная часть молекулы ПАВ
будет обращена к полярной фазе, а
неполярная часть - к неполярной. С
увеличением расстояния от твердой
поверхности ориентация
молекул поверхностно-активного
вещества ПАВ
нарушается, а затем пропадает. Толщина
граничного слоя зависит от строения
молекул и внешних условий. Максимальное
значение краевого угла отвечает полному
насыщению адсорбционного слоя, когда
достигается наиболее
полная предельная ориентация молекул
ПАВ гидрофобными
концами наружу.
Ni +28. 1s2 2s2 2p6 3s2 3p6 3d8 4s2. )1s2)2s2 2p6)3s2 3p6)3d8 4s2. 18Ar[ 3d8 4s2 ]
2 8 16 2
n=1 n=2 n=3 n=4
L=0 L=0,1 L=0,1,2 L=0,1,2,3
me=1 me=1,3 me=1,3,5 me=1,3,5,7
ms= -1/2,+1/2 ms= -1/2,+1/2 ms= -1/2,+1/2 ms= -1/2,+1/2
Физические и химические свойства: никель — ковкий и пластичный металл (степень окисления +2,+3). На воздухе компактный никель стабилен, а высокодисперсный никель пирофорен. Он существует в двух полиморфных модификациях: низкотемпературной и высокотемпературной. Известны такие растворимые в воде соли никеля, как сульфат NiSO4, нитрат Ni(NO3)2 и многие другие. Большинство этих солей при кристаллизации из водных растворов образует кристаллогидраты, например, NiSO4·7Н2О, Ni(NO3)2·6Н2О. К числу нерастворимых соединений никеля относятся фосфат Ni3(PO4)2 и силикатNi2SiO4.
Ni(NO3)2 + 2NaOH = Ni(OH)2 + 2NaNO3 выпадает зеленый осадок гидроксида никеля
Если на суспензию Ni(OH)2 в щелочной среде воздействовать сильным окислителем бромом, то возникает гидроксид никеля (III): 2Ni(OH)2 + 2NaOH + Br2 = 2Ni(OH)3 + 2NaBr
Нахождение в природе: в земной коре содержание никеля составляет около 8·10–3% по массе. Земля примерно на 3 % состоит из никеля, а среди составляющих планету элементов никель занимает пятое место — после железа, кислорода, кремния и магния. Никель содержится в некоторых метеоритах, которые по составу представляют собой сплав никеля и железа.
Биологическая роль: никель относится к числу микроэлементов, необходимых для нормального развития живых организмов. Никель принимает участие в ферментативных реакциях у животных и растений. Повышенное содержание никеля в почвах приводят к эндемическим заболеваниям — у растений появляются уродливые формы, у животных — заболевания глаз, связанные с накоплением никеля в роговице.
Углеводоро́ды — органические соединения, состоящие исключительно из атомов углерода и водорода. Углеводороды считаются базовыми соединениями органической химии, все остальные органические соединения рассматривают как их производные. Галогенирование алканов протекает по радикальному механизму. Галогенирование — это одна из реакций замещения. В первую очередь галогенируется наименее гидрированый атом углерода (третичный атом, затем вторичный, первичные атомы галогенируются в последнюю очередь). Галогенирование алканов проходит поэтапно — за один этап замещается не более одного атома водорода:
CH4 + Cl2 → CH3Cl (хлорметан) + HCl
CH3Cl + Cl2 → CH2Cl2 (дихлорметан) + HCl
CH2Cl2 + Cl2 → CHCl3 (трихлорметан) + HCl
CHCl3 + Cl2 → CCl4 (тетрахлорметан) + HCl.
Моносахариды – гетерофункциональные соединения, в состав их молекул входит одна карбонильная группа (альдегидная или кетонная) и несколько гидроксильных. Общая формула моносахаридов — СnН2nОn (n = 3 — 9). По числу атомов углерода в молекуле моносахариды делятся на триозы (n = 3), тетрозы (n = 4), пентозы (n =5), гексозы (n = 6) и т. д. В природе чаще всего встречаются пентозы и гексозы.
Билет 22
Адсорбция
– процесс самопроизвольного изменения
концентрации равновесного вещества
на границе раздела фаз. Поглощаемое
вещество, ещё находящееся в объёме
фазы, называют адсорбтив,
поглощённое — адсорбат.
В более узком смысле под адсорбцией
часто понимают поглощение примеси из
газа или жидкости твёрдым веществом
или жидкостью — адсорбентом.
При этом, как и в общем случае адсорбции,
происходит концентрирование примеси
на границе раздела адсорбент-жидкость
либо адсорбент-газ. Процесс, обратный
адсорбции, то есть перенос вещества с
поверхности раздела фаз в объём фазы,
называется десорбция.
Если скорости адсорбции и десорбции
равны, то говорят об установлении
адсорбционного
равновесия.
Адсорбция является частным случаем
сорбции, процесс, обратный адсорбции
– десорбция. На поверхности раздела
двух фаз помимо адсорбции, обусловленной
в основном физическими взаимодействиями
может идти химическая реакция. Этот
процесс называется хемосорбцией.
Чёткое разделение на адсорбцию и
хемосорбцию не всегда возможно. Одним
из основных параметров по которым
различаются эти явления является
тепловой эффект: так, тепловой эффект
физической адсорбции обычно близок к
теплоте сжижения адсорбата, тепловой
эффект хемосорбции значительно выше.
Кроме того в отличие от адсорбции
хемосорбция обычно является необратимой
и локализованной. Свободная
энергия Гиббса —
это величина, показывающая изменение
энергии в ходе химической реакции и
дающая таким образом ответ на вопрос
о принципиальной возможности протекания
химической реакции;
![]() Г-кол-во
адсорбированного вещества; С-молярная
концентрация растворенного вещества;
R-универсальная
газовая постоянная
Г-кол-во
адсорбированного вещества; С-молярная
концентрация растворенного вещества;
R-универсальная
газовая постоянная
S+16. 1s 22s 22p 63s 23p4. )1s2)2s2 2p6)3s2 3p4. 10Ne[3s2 3p4] Валентность -2, +4, +6.
2 8 6
n=1 n=2 n=3
L=0 L=0,1 L=0,1,2
me=1 me=1,3 me=1,3,5
ms= -1/2,+1/2 ms= -1/2,+1/2 ms= -1/2,+1/2
Распространение Серы в природе. Сера относится к весьма распространенным химическим элементам встречается в свободном состоянии (самородная сера) и в виде соединений - сульфидов, полисульфидов, сульфатов. Вода морей и океанов содержит сульфаты натрия, магния, кальция. Многие процессы биосферы приводят к концентрации Серы - она накапливается в гумусе почв, углях, нефти, морях и океанах, подземных водах, в озерах и солончаках.
Ф изические
свойства Серы. Сера
- твердое кристаллическое вещество,
устойчивое в виде двух аллотропических
модификаций. Сера - плохой проводник
тепла и электричества. В воде она
практически нерастворима, хорошо
растворяется в безводном аммиаке,
сероуглероде и в ряде органических
растворителей (фенол, бензол, дихлорэтан
и других).
изические
свойства Серы. Сера
- твердое кристаллическое вещество,
устойчивое в виде двух аллотропических
модификаций. Сера - плохой проводник
тепла и электричества. В воде она
практически нерастворима, хорошо
растворяется в безводном аммиаке,
сероуглероде и в ряде органических
растворителей (фенол, бензол, дихлорэтан
и других).
Химические свойства Серы. Конфигурация внешних электронов атома S 3s2Зр4. Сера химически активна и особенно легко при нагревании соединяется почти со всеми элементами, за исключением N2, I2, Au, Pt и инертных газов. С О2 на воздухе выше 300 °С образует оксиды: SO2 - сернистый ангидрид и SO3- серный ангидрид, из которых получают соответственно сернистую кислоту и серную кислоту, а также их соли сульфиты и сульфаты. Уже на холоду S энергично соединяется с F2, при нагревании реагирует с Cl2; с бромом Сера образует только S2Br2, иодиды серы неустойчивы. Na2S + (n–1)S = Na2Sn; Na2SO3 + S = Na2S2O3; Н2О + SO2 = H2SO3; SO3 + H2O = H2SO4
Применение Серы. В медицинской практике применение Серы основано на ее способности при взаимодействии с органических веществами организма образовывать сульфиды и пентатионовую кислоту, от присутствия которых зависят кератолитические, противомикробные и противопаразитарные эффекты. Сера входит в состав мази Вилькинсона и других препаратов, применяемых для лечения чесотки. Очищенную и осажденную Серу употребляют в мазях и присыпках для лечения некоторых кожных заболеваний (себорея, псориаз и других); в порошке - при глистных инвазиях (энтеробиоз); в растворах - для пиротерапии прогрессивного паралича и других.
Сера в организме. В виде органических и неорганических соединений Сера постоянно присутствует во всех живых организмах и является важным биогенным элементом. Ее среднее содержание в расчете на сухое вещество составляет: в морских растениях около 1,2%, наземных - 0,3%, в морских животных 0,5-2%, наземных - 0,5%. Биологическая роль Серы определяется тем, что она входит в состав широко распространенных в живой природе соединений: аминокислот (метионин, цистеин), и следовательно белков и пептидов; коферментов (кофермент А, липоевая кислота), витаминов (биотин, тиамин), глутатиона и других. Неорганические соединения Сера в организмах высших животных обнаружены в небольших количествах, главным образом в виде сульфатов (в крови, моче), а также роданидов (в слюне, желудочном соке, молоке, моче). Животные усваивают Серу в составе органических соединений.
Первичная структура белка - химическая формула белка, представляющая определенную линейную последовательность соединения аминокислот в полипептидной цепи. Гидролиз белков — это необратимое разрушение первичной структуры в кислом или щелочном растворе с образованием аминокислот. У каждого организма имеются белки, свойственные только ему. Каждый белок состоит из 20 различных а-аминокислот. Общая формула аминокислот. R-CHNH2-COOH. Вторичная структура — конформационное расположение главной цепи макромолекулы независимо от конформации боковых цепей или отношения к другим сегментам. В описании вторичной структуры важным является определение водородных связей, которые стабилизируют отдельные фрагменты макромолекул. Это пространственная структура, образующаяся в результате взаимодействия между функциональными группами пептидного остова. а) α-спирали — плотные витки вокруг длинной оси молекулы. Спираль построена исключительно из одного типа аминокислот. Хотя она может быть как левозакрученной, так и правозакрученной, в белках преобладает правозакрученная. б) β-листы — несколько зигзагообразных полипептидных цепей, в которых водородные связи образуются между относительно удалёнными друг от друга в первичной структуре аминокислотами или разными цепями белка, а не близко расположенными, как имеет место в α-спирали. в) π-спирали; 310-спирали;неупорядоченные фрагменты. Третичное-пространственное строение полипептидной цепи. Структурно состоит из элементов вторичной структуры, в которых гидрофобные взаимодействия играют важнейшую роль. В стабилизации третичной структуры принимают участие:1)ковалентные связи;2)ионные связи между противоположно заряженными боковыми группами аминокислотных остатков;3)водородные связи;4)гидрофильно-гидрофобные взаимодействия. Четвертичная - взаимное расположение нескольких полипептидных цепей в составе единого белкового комплекса. Белковые молекулы, входящие в состав белка с четвертичной структурой, образуются на рибосомах по отдельности и лишь после окончания синтеза образуют общую структуру. В состав белка с четвертичной структурой могут входить как идентичные, так и различающиеся полипептидные цепочки.
Химические превращения, в результате которых структурные изомеры превращаются друг в друга, называется изомеризацией. Такие процессы имеют важное значение в промышленности. Так, например, проводят изомеризацию нормальных алканов в изоалканы для повышения октанового числа моторных топлив; изомеризуют пентан в изопентан для последующего дегидрирования визопрен. π-диастереомеры, называемые также геометрическими изомерами, отличаются друг от друга различным пространственным расположением заместителей относительно плоскости двойной связи (чаще всего С=С и С=N) или цикла. К ним относятся, например,малеиновая и фумаровая кислоты (формулы XIV и XV соответственно), (Е)- и (Z)-бензальдоксимы (XVI и XVII), цис- и транс-1,2-диметилциклопентаны (XVIII и XIX).
Билет №23
1) Дисперсные системы
Если одно вещество,находящееся в раздробленном состоянии,равномерно распределено в массе другого вещества,то такую систему наз дисперсной.Раздробленное вещество наз дисперсной фазой ,а среду дисперсной средой
Грубодисперсные системы.Эти системы содержат в качестве дисперсной фазы наиболее крупные частицы диаметром от 0,1мк и выше.К ним относят суспензии и эмульсии.
Суспензиями наз системы,в которых твердое вещество находится в жидкой дисперсионной среде. Эмульсиями наз дисперсные системы двух несмешивающихся жидкостей,где одна жидкость в виде капелек диспергирована в другой
Каллоидные системы. Они имеют размеры частиц дисперсной фазы от 0,1мк до 1мк.Каллоидные частицы при наличии у них электрического заряда и сольватно-ионных оболочек остаются во взвешенном состоянии и очень долго могут не выпадать в осадок.Примером могут быть растворы альбумина,желатина,каллоидные растворы золота.
2) Железо - это d- элемент VIII группы; порядковый номер – 26; атомная масса Ar(Fe) = 56; состав атома: 26-протонов; 30 – нейтронов; 26 – электронов.
формула: 1s22s22p63s23p63d64s2
Металл средней аСхема строения атома:
+26 )2е )8е )14е )2е
Электронная ктивности, восстановитель:
Fe0-2e-→Fe+2, окисляется восстановитель
Fe0-3e-→Fe+3, окисляется восстановитель
Основные степени окисления: +2, +2
Fe0
Fe2+
Fe3+
5. Получение железа
Восстановлением из оксидов углём или оксидом углерода (II), а также водородом:
FeO + C = Fe + CO
Fe2O3 + 3CO = 2Fe + 3CO2
Fe2O3 + 3H2 = 2Fe + 3H2O
6. Химические свойства железа
Как элемент побочной подгруппы железо может проявлять несколько степеней окисления. Мы рассмотрим только соединения, в которых железо проявляет степени окисления +2 и +3. Таким образом, можно говорить, что у железа имеется два ряда соединений, в которых оно двух- и трехвалентно.
1) На воздухе железо легко окисляется в присутствии влаги (ржавление):
4Fe + 3O2 + 6H2 O = 4Fe(OH)3
3) При высокой температуре (700–900°C) железо реагирует с парами воды:
3Fe + 4H2O t˚C→ Fe3O4 + 4H2
4) Железо реагирует с неметаллами при нагревании:
2Fe + 3Br2 t˚C→ 2FeBr3
Fe + S t˚C→ FeS
5) Железо легко растворяется в соляной и разбавленной серной кислотах при обычных условиях:
Fe + 2HCl = FeCl2 + H2
Fe + H2SO4(разб.) = FeSO4 + H2
Оксид Fe03 (кислотный), где степень окисления железа равна +6, непрочен и для медицины не имеет значения.
Оксид FeO, где степень окисления железа равна +2, имеет основной характер.
Fe203 является амфотерным. Два последних оксида FeO и Fe203~ дают ряд солей, которые применяются в медицине.Различают соли железа закисные, где степень окисления железа равна +2, и окисные, где степень окисления железа равна +3
Железо является составной частью нашего организма, большая часть его содержится в гемоглобине крови.Применение препаратов железа в медицине основано на их вяжущем и прижигающем действии в зависимости от концентрации. Закисное железо является физиологически более активным и играет более важную роль в организме, чем окисное. Препараты железа применяются главным образом при анемии, малокровии, а также как кровоостанавливающее средство.
3) Моносахариды,Глюкоза (С6Н12О6)-это простейшие углеводы,являются альдегидами или кетонами,содержат несколько гидросильных групп и соответственно называются альдозами и кетозами
ХОУОРСА ФОРМУЛЫ (Хеуорса ф-лы), изображение на плоскости пространств. структур циклич. соед. При построении Хоуорса формул цикл условно считают плоским (на самом деле молекула м. б. в конформации кресла или ванны) и проецируют на плоскость под нек-рым углом; при этом ближняя к наблюдателю часть кольца на чертеже располагается снизу и обычно выделяется более жирной линией (рис.). В моносахаридах кислородный атом цикла располагают обычно на наиб. удалении от наблюдателя (в случае пиранозного цикла - справа).
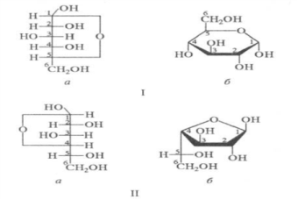
Цикло-оксо таутомерия
В кристаллическом состоянии все
моносахариды находятся в циклической
форме в виде α- или β-аномеров. При
растворении моносахаридов в воде
наблюдается так называемая цикло-оксо
таутомерия (кольчато-цепная таутомерия),
т.е. устанавливается равновесие между
циклическими таутомерами моносахаридов,
различающихся по размеру цикла
(фуранозный, пиранозный), α- и β-формами
и открытой формой. Существование
равновесия между линейной и циклическими
формами моносахаридов получило
названиецикло-оксо-таутомерии.
кристаллическом состоянии все
моносахариды находятся в циклической
форме в виде α- или β-аномеров. При
растворении моносахаридов в воде
наблюдается так называемая цикло-оксо
таутомерия (кольчато-цепная таутомерия),
т.е. устанавливается равновесие между
циклическими таутомерами моносахаридов,
различающихся по размеру цикла
(фуранозный, пиранозный), α- и β-формами
и открытой формой. Существование
равновесия между линейной и циклическими
формами моносахаридов получило
названиецикло-оксо-таутомерии.
В циклической форме возникает дополнительный центр хиральности, т.е. асимметрическим становится карбонильный атом углерода. Этот новый хиральный центр называют аномерным, а два стереоизомера – α- и β-аномеры, ОН – называют гликозидный гидроксил.
4) Карбонилами называют соединения, содержащие атомы металлов и молекулы окиси углерода. Образование карбонилов характерно только для переходных металлов, по-видимому, потому, что только атомы переходных металлов способны вступать одновременно в донорно-акцепторную и я-дативную связь с окисью углерода. Исключение составляют цирконий и гафний, которые карбонильных. Соединений не образуют.
Карбонилы родственные металлорганическим соединениям (МОС) . Они также содержат связь М—С. Многие из них, как и МОС, легколетучи и обладают значительным давлением пара часто даже при комнатной температуре. Они, как правило, растворяются в неполярных растворителях, а в полярных, например в воде, нерастворимы. В термическом отношении карбонилы неустойчивы: термолиз с разложением на металл и СО наступает при нагревании до температуры выше 200°С. Это делает карбонилы очень удобными для ряда технических и технологических целей. Карбонилы металлов, соединения металлов с окисью углерода общей формулы Mem(CO)n. Впервые (в 1890) был открыт карбонил никеля Ni(CO)4. С тех пор получены карбонилы многих металлов и некоторых неметаллов. В зависимости от числа атомов металла в молекуле К. м. могут быть "одноядерными" и "многоядерными"; известны также смешанные К. м., например [Co(CO)4]2Zn.
Карбоксилат-ион - С-О - является акцептором протона. Карбоксилат-ион построен симметрично и имеет систему сопряженных связей. Если карбоксилат-ион может быть стабилизирован внутримолекулярными водородыыми связями, это приведет к возрастанию кислотности родо-начальной карбоновой кислоты. В качестве примера ниже сравниваются величины кислотности салициловой кислоты ( о-оксибензойной кислоты), для которой возможно образование внутримолекулярных водородных связей, и о-метоксибензойной кислоты, для которой это исключено. Из карбоксилат-иона получается енолят, а свободная кислота дает енол ацетона. Алкилирование карбоксилат-ионов катализируется также краун-эфирами. Обычно реакция проводилась с алкилгалогени-дом в органической фазе, например в ацетонитриле, который находится в контакте с твердой солью карбоновой кислоты. Сочетание краун-эфира и растворителя ( по крайней мере довольно полярных растворителей, подобных ацетонитрилу) способствует растворению соли. Слабо сольватированный карбо-ксилат-анион - эффективный нуклеофил в таких условиях, поэтому алкилирование происходит легко. Ионная пара ацетат калия - краун-эфир является реакционноспособным нуклеофи-лом, но, вероятно, относительно слабым основанием. Стабильность карбоксилат-иона обусловлена тем, что отрицательный заряд равномерно распределяется на оба атома кислорода. Поэтому точнее изображать карбоксилат-ион так, как будто каждый атом кислорода несет на себе половинный заряд и связан с атомом углерода равноценными связями, являющимися промежуточными между простой и двойной связью. Подобно карбоксилат-ионам и имидазольным группам серусодержащие группы могут образовывать координационные комплексы с ионами металлов. По-видимому, серусодержащие остатки аминокислот мало участвуют в образовании водородных связей между боковыми цепями. В карбоксилат-ионе заряд делокализуется по сопряженной системе с участием двух атомов кислорода. Описание этого явления с привлечением резонансных структур показывает, что отрицательный заряд не принадлежит какому-либо одному атому кислорода, а равномерно распределен между ними. Такое строение кар-боксилат-иона доказано с помощью рентгеноструктурного анализа. Как и карбоксилат-ион, анион карбоната GOJ стабилизован за счет резонанса, что обычно изображают при помощи обоюдоострых стрелок между реально несуществующими предельными структурами. На примере карбоксилат-иона в качестве заместителя впервые было обнаружено, что влияние соседней группы может определять стереохимию нуклеофильного замещения. Это верно и для присоединения.
Билет№24
1) Строение коллоидной мицеллы
Лиофобные коллоиды обладают очень высокой поверхностной энергией и являются поэтому термодинамически неустойчивыми; это делает возможным самопроизвольный процесс уменьшения степени дисперсности дисперсной фазы (т.е. объединение частиц в более крупные агрегаты) – коагуляцию золей. Тем не менее золям присуща способность сохранять степень дисперсности – агрегативная устойчивость, которая обусловлена, во-первых, снижением поверхностной энергии системы благодаря наличию на поверхности частиц дисперсной фазы двойного электрического слоя и, во-вторых, наличием кинетических препятствий для коагуляции в виде электростатического отталкивания частиц дисперсной фазы, имеющих одноименный электрический заряд.Строение структурной единицы лиофобных коллоидов – мицеллы – может быть показано лишь схематически, поскольку мицелла не имеет определенного состава. Рассмотрим строение коллоидной мицеллы на примере гидрозоля иодида серебра, получаемого взаимодействием разбавленных растворов нитрата серебра и иодида калия:
AgNO3 + KI ––> AgI + KNO3
Коллоидная мицелла золя иодида серебра (см. рис. 4.9) образована микрокристаллом иодида серебра, который способен к избирательной адсорбции из окружающей среды катионов Ag+ или иодид-ионов. Если реакция проводится в избытке иодида калия, то кристалл будет адсорбировать иодид-ионы; при избытке нитрата серебра микрокристалл адсорбирует ионы Ag+. В результате этого микрокристалл приобретает отрицательный либо положительный заряд; ионы, сообщающие ему этот заряд, называются потенциалопределяющими, а сам заряженный кристалл – ядром мицеллы. Заряженное ядро притягивает из раствора ионы с противоположным зарядом – противоионы; на поверхности раздела фаз образуется двойной электрический слой. Некоторая часть противоионов адсорбируется на поверхности ядра, образуя т.н. адсорбционный слой противоионов; ядро вместе с адсорбированными на нем противоионами называют коллоидной частицей или гранулой. Остальные противоионы, число которых определяется, исходя из правила электронейтральности мицеллы, составляют диффузный слой противоионов; противоионы адсорбционного и диффузного слоев находятся в состоянии динамического равновесия адсорбции – десорбции.
Схематически мицелла золя иодида серебра, полученного в избытке иодида калия (потенциалопределяющие ионы – анионы I–, противоионы – ионы К+) может быть изображена следующим образом: {[AgI]m · nI– · (n-x)K+}x– · x K+
При получении золя иодида серебра в избытке нитрата серебра коллоидные частицы будут иметь положительный заряд: {[AgI]m · nAg+ · (n-x)NO3–}x+ · x NO3–
2) АЗОТ (лат. Nitrogenium — рождающий селитры), N (читается «эн»), химический элемент второго периода VA группы периодической системы, атомный номер 7, атомная масса 14,0067. В свободном виде — газ без цвета, запаха и вкуса, плохо растворим в воде. Состоит из двухатомных молекул N2, обладающих высокой прочностью. Относится к неметаллам.
Природный азот состоит из стабильных нуклидов 14N (содержание в смеси 99,635% по массе) и 15N. Конфигурация внешнего электронного слоя 1s22s22р3. Радиус нейтрального атома азота 0,074 нм, радиус ионов: N3– — 0,132, N3+ — 0,030 и N5+ — 0,027 нм. Энергии последовательной ионизации нейтрального атома азота равны, соответственно, 14,53, 29,60, 47,45, 77,47 и 97,89 эВ. По шкале Полинга электроотрицательность азота 3,05.
Азот немного легче воздуха; плотность 1,2506 кг/м3 ( при 00С и 101325 н/м2 или 760 мм. рт. ст. ), tпл-
209,860С, tкип-195,80С. Азот сжижается с трудом: его критическая температура довольно низка (-147,10С), а критическое давление высоко
3,39 Мн/м2 (34,6 кгс/см2);плотность жидкого азота 808 кг/м3. В воде азот менее растворим, чем кислород: при 00С в 1 м3 H2O растворяется 23,3 г азота. Лучше, чем в воде, азот растворим в некоторых углеводородах.
Только с такими активными металлами, как литий, кальций, магний, азот взаимодействует при нагревании до сравнительно невысоких температур. С большинством других элементов азот реагирует при высокой температуре и в присутствии катализаторов. Хорошо изучены соединения азота с кислородом
N2O, NO, N2O3, NO2 и N2O5. Из них при непосредственном взаимодействии элементов ( 40000С ) образуется окись NO, которая при охлаждении легко окисляется далее до двуокиси NO2. В воздухе окислы азота образуются при атмосферных разрядах. Их можно получить также действием на смесь азота с кислородом ионизирующих излучений. При растворении в воде азотистого N2O3 и азотного N2O5 ангидридов соответственно получаются азотистая кислота
НNO2 и азотная кислота НNO3, образующие соли - нитриты и нитраты. С водородом азот соединяется только при высокой температуре и в присутствии катализаторов, при этом образуется аммиак NH3.
В медицине для сохранения крови и кровесодержащих препаратов, для быстрого замораживания и хранения тканей и различных органов, в технологиях получения полноценных порошковых лекарственных препаратов, в криотерапии.
3) Жирные кислоты подразделяются на предельные (насыщенные) и непредельные (ненасыщенные). Наиболее распространены насыщенные жирные кислоты — пальмитиновая, стеариновая, масляная и капроновая. Пальмитиновая и стеариновая кислоты — высокомолекулярные и являются твердыми веществами.Жирные кислоты содержат в своей молекуле кислотную группу —СООН (карбоксильную группу). «Жирными» их называют потому, что некоторые высокомолекулярные члены этого ряда входят в состав жиров.
Общая формула жирных кислот имеет вид R-COOH, где R — атом водорода или радикал типа —СН3, —С2Н5 и т. д.; каждый следующий член этого ряда отличается от предыдущего на одну группу СН2.
Насыщенные жирные кислоты содержатся в жирах животного происхождения. Они обладают невысокой биологической активностью и могут оказывать отрицательное действие на жировой и холестериновый обмены.
Ненасыщенные жирные кислоты широко представлены во всех пищевых жирах, но больше всего их находится в растительных маслах. Они содержат двойные ненасыщенные связи, что обусловливает их значительную биологическую активность и способность к окислению. Самыми распространенными являются олеиновая, линолевая, линоленовая и арахидоновая жирные кислоты, среди которых наибольшей активностью обладает арахидоновая кислота.
Ненасыщенные жирные кислоты в организме не образуются и должны ежедневно вводиться с пищей в количестве 8— 10 г. Источниками олеиновой, линолевой и линоленовой жирных кислот являются растительные масла. Арахидоновая жирная кислота почти не содержится ни в одном продукте и может синтезироваться в организме из линолевой кислоты в присутствии витамина В6 (пиридоксина).
Недостаток ненасыщенных жирных кислот приводит к задержке роста, возникновению сухости и воспалению кожных покровов.
Ненасыщенные жирные кислоты входят в состав мембранной системы клеток, миелиновых оболочек и соединительной ткани. Известно участие их в жировом обмене и в переводе холестерина в легкорастворимые соединения, которые выводятся из организма.
Насыщенные жирные кислоты (НЖК), наиболее представленные в пище, делятся на короткоцепочечные (4… 10 атомов углерода — масляная, капроновая, каприловая, каприновая), среднецепочечные (12… 16 атомов углерода — лауриновая, миристиновая, пальмитиновая) и длинноцепочечные (18 атомов углерода и более — стеариновая, арахидиновая).
4) Алкалоиды - это природные азотсодержащие органические соединения основного характера, имеющие сложный состав и обладающие сильным специфическим действием. Большинство их относится к соединениям с гетероциклическим атомом азота в кольце, реже азот находится в боковой цепи. Синтезируются преимущественно растениями. В переводе термин "алкалоид" (от араб. "alkali" - щелочь и греч. "eidos" - подобный) означает щелочноподобный. Подобно щелочам, алкалоиды образуют с кислотами соли.
Распространение. В растительном мире распределены неравномерно. В низших растениях их мало. Встречаются в семействе плауновых (плаун-баранец). У злаков и осоковых растений встречаются редко. Наиболее богаты алкалоидами растения семейств маковых, пасленовых, лилейных, мареновых, сельдерейных, амариллисовых, бобовых, лютиковых. В растениях алкалоиды находятся в клеточном соке в растворенном виде. Содержание колеблется от тысячных долей процента до нескольких процентов, а в коре хинного дерева от 15 до 20%.
У некоторых растений алкалоиды содержатся во всех органах (красавка обыкновенная и кавказская), у большинства они преобладают в каком-либо одном органе. Часто у одного растения в разных органах имеется различное число алкалоидов, некоторые органы могут быть безалкалоидными, например) мак опийный во всех органах, кроме семян, содержит алкалоиды. Обычно в растении встречается несколько алкалоидов: в опии, например, 26 алкалоидов, в корнях раувольфии - 35. Редко присутствует в растении один алкалоид.
Физико-химические свойства.
В состав алкалоидов в основном входят углерод, водород, азот и кислород; алкалоиды кубышки дополнительно содержат серу. Большинство алкалоидов, содержащих кислород - бесцветные, оптически активные, кристаллические или аморфные вещества со щелочной реакцией; некоторые алкалоиды окрашены (например, алкалоид берберин из барбариса желтого цвета), без запаха, горького вкуса. Бескислородные алкалоиды - летучие жидкости с неприятным запахом (например, алкалоид никотин из табака, кониин из болиголова).
Алкалоиды-основания, в воде почти нерастворимы; растворяются в спирте, эфире, хлороформе и других органических растворителях. Соли алкалоидов растворимы в воде и спирте, но нерастворимы в органических растворителях. Алкалоиды в растениях находятся в виде солей, связаны с органическими кислотами: щавелевой, лимонной, яблочной, винной. Для мака снотворного характерна меконовая кислота, а для хинной коры - хинная кислота.
Кофеин как химическое вещество принадлежит к группе ксантинов. К этой же группе принадлежит аденозин, который естественным образом присутствует в головном мозге и выступает в качестве нейромедиатора в ряде синапсов. Один из эффектов кофеина связан с его вмешательством в те процессы в различных местах головного мозга, которые регулируются аденозином, в том числе в так называемой ретикулярной формации, отвечающей за уровень бодрствования организма.
Когда аденозин связывается с соответствующими рецепторами, это приводит к замедлению деятельности и вызывает чувство усталости. Поскольку кофеин имеет сходную с аденозином структуру, он связывается с теми же рецепторами, но при этом не происходит снижения активности клеток, наоборот, она возрастает. Кроме того, кровеносные сосуды головного мозга суживаются.
Организм в ответ на эту возросшую активность клеток головного мозга действует так, как будто головной мозг зафиксировал возникновение опасности. Гипофиз выделяет гормон, который заставляет надпочечники синтезировать больше адреналина. Адреналин заставляет сердце сокращаться чаще, а печень выбрасывать в кровоток больше глюкозы, чтобы организм мог использовать дополнительное количество энергии. Кроме того, кофеин суживает кровеносные сосуды, расслабляет дыхательные пути, что облегчает дыхание, и позволяет мышцам сокращаться с большей легкостью. Организм мобилизован для активной деятельности.
Кроме увеличения энергии, кофеин оказывает другие виды действия. Он увеличивает мочеотделение и подавляет аппетит.
Кофеин является стимулянтом центральной нервной системы. В умеренных дозах кофеин может усиливать состояние бодрствования, затруднять координацию тонких движений, изменять стереотипы сна, а именно увеличивать необходимое для засыпания время и сокращать общее время сна, вызывать головные боли, нервозность и головокружение. Кофеин может вызывать тревогу и напряженность
Билет №25
1) Электрокинетические явления
Электорокинетические явления были открыты в 1808 году профессором Московского университета Ф.Ф.Рейссом. Исследуя электролиз воды, он обнаружил, что если мелкий кварцевый песок поместить в среднюю часть U- образной трубки так, что бы он образовал как бы пористую диафрагму, заполнить трубку водой и приложить электрическое поле к электродам. опущенным в об а колена трубки, то уровень воды в колене с отрицательно заряженным электродом уровень воды будет подниматься до тех пор, пока разность уровней в обоих коленах не достигнет определённого значения
Это явление переноса частиц дисперсной фазы в электрическом поле получило название электрофореза или катафореза. Скорость переноса частиц тем выше, чем больше разность потенциалов между электродами, диэлектрическая проницаемость среды, и тем меньше, чем больше вязкость дисперсной среды.
Электрофорез
1808 г Ф.Ф.Рейс установил, что при наложении разности электрического потенциала на электроды, опущенные в заполненные водой стеклянные трубки, воткнутые в кусок сырой глины, жидкость в трубке с положительным полюсом мутнела за счет появления там частиц глины. Это указывало на то, что частицы глины переносятся в электрическом поле к положительному полюсу. Перенос частиц в электрическом поле получил название электрофореза. Процесс протекает с постоянной скоростью, зависящей от величины разности потенциалов и характеристик среды:
![]()
где U - линейная скорость движения границы золь-жидкость, м/с;
ε – диэлектрическая проницаемость среды, ф/м;
ε0 – электрическая константа (8,85 × 10-12 ф/м);
Н – градиент внешнего поля, В/м;
η- вязкость среды, Н × с/м2;
ξ – дзета-потенциал.
Электроосмос
Ф.Ф. Рейс открыл также явление электроосмоса, заключающееся в том, что если тонкий порошок кварца поместить в среднюю часть U-образной трубки так, чтобы он образовал род пористой диафрагмы, заполнить трубку водой и приложить электрический ток к электродам, помещенным в оба колена трубки, то уровень воды в колене с отрицательным электродом будет повышаться до тех пор, пока разность уровней в обоих коленах не достигнет определенной величины. Процесс идет с постоянной скоростью, которую можно рассчитать по следующей формуле:
![]()
где υ – объемная скорость, м3/с;
I – сила тока, А;
χ – удельная электропроводность, Ом-1 м-1;
ξ – дзета-потенциал.
Система очищения организма от токсинов, разработанная специалистами центра, показала высокий эффект при многих хронических тяжелых заболеваниях. электрофорез при кисте Предложенные нами методы вывести токсины из организма хорошо себя зарекомендовали при лечении больных облитерирующими заболеваниями нижних конечностей. электроосмос и электрофорез . зависимости от происхождения токсины можно разделить их на два вида - это эндогенные и экзогенные. Вещества первой группы могут быть вызваны различными заболеваниями, среди которых грипп, диабет, простуда, злокачественные образования и многие другие недуги. электроосмос и электрофорез . Вещества второй группы могут быть вызваны применением различных лекарственных и химических средств, используемые человеком в повседневной жизни.
2) Элемент цинк (Zn) в таблице Менделеева имеет порядковый номер 30. Он находится в четвертом периоде второй группы. Атомный вес - 65,37. Распределение электронов по слоям 2-8-18-2. Цинк представляет собой синевато - белый металл, плавящийся при 419 С, а при 913 С превращающийся в пар; плотность его равна 7,14 г/см3. При обыкновенной температуре цинк довольно хрупок, но при 100-110 С он хорошо гнется и прокатывается в листы. На воздухе цинк покрывается тонким слоем окиси или основного карбоната, предохраняющим его от дальнейшего окисления. Вода почти не действует на цинк, хотя он и стоит в ряду напряжений значительно левее водорода. Это объясняется тем, что образующаяся на поверхности цинка при
взаимодействии его с водой гидроокись практически нерастворима и препятствует дальнейшему течению реакции. В разбавленных же кислотах цинк легко растворяется с образованием соответствующих солей. Кроме того, цинк подобно
бериллию и другим металлам, образующим амфотерные гидроокиси, растворяется в щелочах. Если нагреть цинк на воздухе до температуры кипения, то пары его воспламеняются и сгорают зеленовато-белым пламенем, образуя окись цинка.
Физические свойства Цинка. Цинк - металл средней твердости. В холодном состоянии хрупок, а при 100-150 °С весьма пластичен и легко прокатывается в листы и фольгу толщиной около сотых долей миллиметра. При 250 °С вновь становится хрупким. Полиморфных модификаций не имеет. Кристаллизуется в гексагональной решетке с параметрами а = 2,6594Å, с = 4,9370Å. Атомный радиус 1,37Å; ионный Zn2+ -0,83Å. Плотность твердого Цинка 7,133 г/см3 (20 °С), жидкого 6,66 г/см3 (419,5 °С); tпл 419,5 °С; tкип 906 °С. Температурный коэффициент линейного расширения 39,7·10-3 (20-250 °С), коэффициент теплопроводности 110,950 вт/(м ·К) 0,265 кал/см·сек·°С (20 °С), удельное электросопротивление 5,9·10-6 ом·см (20 °С), удельная теплоемкость Цинка 25,433 кдж/(кг·К.) [6,07 кал/(г·°С)]. Предел прочности при растяжении 200-250 Мн/м2 (2000-2500 кгс/см2), относительное удлинение 40-50%, твердость по Бринеллю 400-500 Мн/м2(4000-5000 кгс/см2). Цинк диамагнитен, его удельная магнитная восприимчивость -0,175·10-6. Химические свойства Цинка. Внешняя электронная конфигурация атома Zn 3d104s2. Степень окисления в соединениях +2. Нормальный окислительно-восстановительный потенциал, равный 0,76 в, характеризует Цинк как активный металл и энергичный восстановитель. На воздухе при температуре до 100 °С Цинк быстро тускнеет, покрываясь поверхностной пленкой основных карбонатов. Во влажном воздухе, особенно в присутствии СО2, происходит разрушение металла даже при обычных температурах. При сильном нагревании на воздухе или в кислороде Цинк интенсивно сгорает голубоватым пламенем с образованием белого дыма оксида цинка ZnO. Сухие фтор, хлор и бром не взаимодействуют с Цинком на холоду, но в присутствии паров воды металл может воспламениться, образуя, например, ZnCl2. Нагретая смесь порошка Цинка с серой дает сульфид Цинк ZnS. Сульфид Цинк выпадает в осадок при действии сероводорода на слабокислые или аммиачные водные растворы солей Zn. Гидрид ZnH2 получается при взаимодействии LiАlН4 с Zn(CH3)2 и других соединениями Цинка; металлоподобное вещество, разлагающееся при нагревании на элементы. Нитрид Zn3N2 - черный порошок, образуется при нагревании до 600 °С в токе аммиака; на воздухе устойчив до 750 °С, вода его разлагает. Карбид Цинка ZnC2 получен при нагревании Цинка в токе ацетилена. Сильные минеральные кислоты энергично растворяют Цинк, особенно при нагревании, с образованием соответствующих солей. При взаимодействии с разбавленной НCl и H2SO4 выделяется Н2, а с НNО3 - кроме того, NO, NO2, NH3. С концентрированной НCl, H2SO4 и HNO3 Цинк реагирует, выделяя соответственно Н2, SO2, NO и NO2. Растворы и расплавы щелочей окисляют Цинк с выделением Н2 и образованием растворимых в воде цинкитов. Интенсивность действия кислот и щелочей на Цинк зависит от наличия в нем примесей. Чистый Цинк менее реакционноспособен по отношению к этим реагентам из-за высокого перенапряжения на нем водорода. В воде соли Цинка при нагревании гидролизуются, выделяя белый осадок гидрооксида Zn(OH)2. Известны комплексные соединения, содержащие Цинк, например [Zn(NH3)4]SО4 и другие.
Цинк для нервов – тоже самое, что железо для крови. Он контролирует, управляет некоторыми процессами обмена веществ, функционированием ферментной системы, поддерживает здоровье клеток, участвует в синтезе белков и образования ДНК, участвует в образовании инсулина, поддерживает кислотно-щелочной баланс в крови, участвует в образовании полового гормона, тестостерона и спермы, играет важную роль в работе мозга и является важным средством при нервном и мозговом истощении.
Наш организм способен откладывать про запас только незначительное количество цинка, поэтому мы нуждаемся в его постоянном пополнении.при нехватке цинка только в течении одной недели начинается замедление роста мышечной ткани и ослабление иммунной системы.
3) Пиримидиновые и пуриновые основания
На основе типичных представителей азотистых гетероциклов – пиридина и пиррола – можно рассмотреть соединения, которые содержат более одного гетероатома в молекуле.
Особенности строения оснований пиримидина и пурина:
![]()
1) это бесцветные кристаллические вещества;
2) пиримидин – шестичленный цикл, подобный пиридину, который отличается от него наличием в молекуле еще одного гетероатома (азота) вместо группы СН; 3) пурин является бициклическим.
Особый интерес представляют не столько пиримидин и пурин, сколько вещества с их характерной структурой – пиримидиновые и пуриновые основания, которые входят в состав природных высокомолекулярных веществ – нуклеиновых кислот, которые осуществляют синтез белков в организмах.
Структурные формулы пиримидиновых оснований:
![]()
Цитозин – (2-гидрокси-4-аминопиримидин) – бесцветное малорастворимое вещество с Тпл 320–325 °C. Цитозин является слабым основанием, сравнимым с анилином и очень слабой NH-кислотой. Цитозин входит в состав нуклеиновых кислот. Урацил (2,4-дигидроксипиримидин) – бесцветное малорастворимое в воде вещество с Тпл 335 °C. Входит в состав нуклеиновых кислот, нуклеотидов. Получают из гидролизатов нуклеиновых кислот. Урацил вступает в реакции электрофильного замещения: алкилирования, галогенирования, азосочетания. Тимин (2,4-дигидрокси-5-метилпиримидин) – бесцветное малорастворимое кристаллическое вещество с Тпл 318 °C. Являясь производным урацила, обнаруживает сходные свойства, за исключением реакций SE, поскольку 5-е положение занято метильным радикалом. Входит в состав нуклеиновых кислот, нуклеотидов, является основой лекарственных препаратов. Например, азидотимидин – лекарство против СПИДа.
Структурные формулы пуриновых оснований:
![]()
аденин (6-аминопурин) – бесцветное кристаллическое вещество с Тпл 360–365 °C, мало растворяется в воде. Входит в состав нуклеотидов, нуклеозидов и нуклеиновых кислот. Его используют в качестве исходного соединения для органического и микробиологического синтеза и в медицине, например в качестве консерванта донорской крови. Гуанин (2-амино-6-гидроксипурин) – бесцветное кристаллическое вещество с Тпл 365 °C, мало растворяется в воде, входит в состав нуклеотидов, нуклеозидов и нуклеиновых кислот.
4) Липиды — большая группа веществ биологического происхождения, хорошо растворимых в органических растворителях, таких, как метанол, ацетон, хлороформ и бензол. В то же время эти вещества нерастворимы или мало растворимы в воде. Слабая растворимость связана с недостаточным содержанием в молекулах липидов атомов с поляризующейся электронной оболочкой, таких, как О, N, S или P .Липиды подразделяются на омыляемые и неомыляемые. Из огромного множества липидов здесь приведены лишь некоторые представители. Отдельные классы липидов обсуждаются в последующих разделах.
О мыляемые
липиды. Структурные компоненты омыляемых
липидов связаны сложноэфирной связью.
Эти липиды легко гидролизуются в воде
под действием щелочей или ферментов.
Омыляемые липиды включают три группы
веществ: сложные эфиры, фосфолипиды и
гликолипиды. В группу сложных эфиров
входят нейтральные жиры (глицерин+три
жирные кислоты), воски (жирный спирт+жирная
кислота) и эфиры стеринов (стерин+жирная
кислота). Группа фосфолипидов включает
фосфатидовые кислоты (глицерин+две
жирные кислоты+фосфатная группа),
фосфатиды (глицерин+две жирные
кислоты+фосфатная группа+спирт) и
сфинголипиды (сфингозин+жирная
кислота+фосфатная группа+спирт). К
группе гликолипидов относятся цереброзиды
(сфингозин+жирная кислота+один углеводный
остаток) и ганглиозиды (сфингозин+жирная
кислота+несколько углеводных остатков,
в том числе нейраминовая кислота).
мыляемые
липиды. Структурные компоненты омыляемых
липидов связаны сложноэфирной связью.
Эти липиды легко гидролизуются в воде
под действием щелочей или ферментов.
Омыляемые липиды включают три группы
веществ: сложные эфиры, фосфолипиды и
гликолипиды. В группу сложных эфиров
входят нейтральные жиры (глицерин+три
жирные кислоты), воски (жирный спирт+жирная
кислота) и эфиры стеринов (стерин+жирная
кислота). Группа фосфолипидов включает
фосфатидовые кислоты (глицерин+две
жирные кислоты+фосфатная группа),
фосфатиды (глицерин+две жирные
кислоты+фосфатная группа+спирт) и
сфинголипиды (сфингозин+жирная
кислота+фосфатная группа+спирт). К
группе гликолипидов относятся цереброзиды
(сфингозин+жирная кислота+один углеводный
остаток) и ганглиозиды (сфингозин+жирная
кислота+несколько углеводных остатков,
в том числе нейраминовая кислота).
Реакция гидролиза.Эта реакция свойственна для жиров.В живых организмах она происходит под действием катализатора,а вне организма катализатором яв-ся щелочь и требуется нагревание.Если гидролиз жира происходит под действием щелочи то реакция наз омылением.
Билет №26
1) Обычные коллоидные системы в отличие от молекулярных растворов вследствие наличия поверхности раздела частиц с дисперсионной средой гетерогенны, большей частью термодинамически неравновесны и агрегативно неустойчивы. Именно поэтому проблема устойчивости коллоидных систем является центральной проблемой коллоидной химии, а коагуляция составляет наиболее важный механизм перехода к более устойчивому состоянию для всех типичных коллоидных систем.Слипание коллоидных частиц и Образование из них более сложных агрегатов называется коагуляцией. Неорганические коллоидные системы особенно склонны к коагуляции под действием высокозаряженных электролитов. Влияние различных электролитов на коагуляцию сравнивают, пользуясь величиной порога коагуляции (минимальная концентрация электролита, вызывающая коагуляцию).
В связи с этим агрегативная устойчивость системы обычно означает медленность процесса коагуляции, т. е. носит кинетический, а не термодинамический характер. Даже полное агрегативное равновесие, когда процессы агресации и распада агрегатов взаимно уравновешиваются, еще не означает термодинамического равновесия всей системы в целом.
Коагулирующее действие электролита очень сильно зависит от валентности того из его иОнов, заряд которого противоположен по знаку заряду самих коллоидных частиц. Чем выше валентность подобного иона, тем меньшая концентрация его необходима для достижения седиментации
оагулирующее действие электролита на латексы, стабилизованные НПАВ, существенно отличается от их действия на системы с ионогенным стабилизатором. Согласно правилам коагуляции электролитами, астабилизующее действие на системы оказывает в основном ион, одноименный по знаку заряда с противоионом двойного электрического слоя. На латексы с неионогенным стабилизатором наибольшее коагулирующее влияние оказывает анион электролита, поскольку анион сильнее дегидратирует неионогенное вещество в адсорбционном слое, разрушая водородные связи между молекулами воды и. Степень дегидратации НПАВ электролитом определяется положением аниона в лиотропном ряду."Движущей силой" коагуляции является избыточная поверхностная энергия. Однако избыток поверхностной энергии мицелл гидрофобного золя частично компенсируется наличием двойных электрических слоев(ДЭС). Электрические силы обусловливают отталкивание одноименно заряженных поверхностей, препятствуя их слипанию; поэтому нарушения строения ДЭС и тем более снижение заряда поверхности до нуля должны снижать агрегативную устойчивость гидрозоля.
Экспериментально установленные закономерности при коагуляцииэлектролитами известны под названием правил коагуляции:
коагуляцию вызывают любые электролиты, но с заметной скоростью она начинается лишь при достижении определенной концентрации; минимальная концентрация электролита, при превышении которой наблюдается коагуляция, называется "порогом коагуляции";2) коагулирующим действием обладает лишь тот ион электролита,заряд
которого противоположен заряду коллоидной частицы, причем его коагулирующая способность тем сильнее, чем выше валентность; коагу-
лирующее действие 2) двухзарядных ионов обычно выше в ≅10-80 раз, а трехзарядных – в ≅350-1500, чем ионов однозарядных. Эта закономерность называется правилом Шульце-Гарди, так как она впервые была установлена Шульце в 1882 г. и дополнена Гарди в 1900 г.;3) в ряду органических ионов коагулирующее действие возрастает с повышением адсорбционной способности; 4) в ряду неорганических ионов с одинаковым зарядом их коагули- рующая активность возрастает с уменьшением гидратации; например, в ряду однозарядных катионов и анионов коагулирующая активность и гидратация изменяются следующим образом:возрастание коагулирующей активности возрастание степени гидратации. Подобные ряды, в которых располагаются ионы одинакового заряда по уменьшению степени гидратации, называются лиотропными рядами
или рядами Гофмейстера; 5) началу коагуляции обычно соответствует снижение ζ –потенциала до критической величины (около 0.03 В); 6) в осадках, получаемых при электролитной коагуляции, всегда присутствуют ионы, вызывающие ее; например, при коагуляции хлоридом бария золя сульфида мышьяка, частицы которого имеют отрицательный заряд, в осадке содержится некоторое количество Ba2+.
2) Медь (лат. Cuprum), Сu, d элемент находится в 4 большом периоде I группе побочной подгруппе периодической системы Менделеева; атомный номер 29, атомная масса 63,546; мягкий, ковкий металл красного цвета. Природная Медь состоит из смеси двух стабильных изотопов - 63Сu (69,1%) и 65Сu (30,9%). (Ar)3d104s1.+29 )2)8)18)1.Валентность 1 и 2.
Медь входит более чем в 198 минералов, из которых для промышленности важны только 17, преимущественно сульфидов, фосфатов, силикатов, карбонатов, сульфатов. Главными рудными минералами являются халькопирит CuFeS2, ковеллин CuS, борнит Cu5FeS4, халькозин Cu2S. Окислы: тенорит, куприт. Карбонаты: малахит, азурит. Сульфаты: халькантит, брошантит. Сульфиды: ковеллин, халькозин, халькопирит, борнит. Эти же цвета, характерны и для многих соединений меди, как в твердом состоянии, так и в растворах. Понижение окраски при повышении валентности видно из следующих двух примеров: CuCl - белый, Cu2O - красный, CuCl2+H2O - голубой, CuO - черный. Карбонаты характеризуются синим и зеленым цветом при условии содержания воды. Практическое значение имеют: самородная медь, сульфиды, сульфосоли и карбонаты (силикаты). Химические свойства: неактивный металл; в ряду напряжений стоит после водорода. Медь устойчива при обычной температуре в сухом и влажном воздухе. Не реагирует с кислотами-неокислителями. Взаимодействует при нагревании с концентрированной серной кислотой и при обычной температуре с разбавленной и концентрированной азотной кислотой. При нагревании реагирует с неметаллами.Применение меди в медицине. В медицине медь в виде сульфата меди также применяется в качестве антисептического и вяжущего средства в виде глазных капель при конъюнктивитах и глазных карандашей для лечения трахомы. Раствор сульфата медь используют также при ожогах кожи фосфором. Иногда сульфат меди применяют как рвотное средство. Нитрат меди употребляют в виде глазной мази при трахоме и конъюнктивитах.
N. 27
1. ГРУБОДИСПЕРСНЫЕ СИСТЕМЫ
Дисперсные системы, размер частиц в которых более 10-5 см называются грубодисперсными. К ним относят эмульсии, суспензии, порошки, пены. В таких системах коллоидные частицы можно увидеть невооруженным глазом и их растворы, как правило, не прозрачные. В грубодисперсных системах практически отсутствует броуновское движение, поэтому они кинетически неустойчивы. Частицы дисперсной фазы в них сравнительно быстро оседают или всплывают. Многие пищевые продукты относят к этой группе дисперсных систем – молоко, майонез, сливочное масло (эмульсии); суфле, муссы, пастила, зефир, хлеб (пены); мука, крахмал, какао (порошки); суспензия крахмала в воде, протертые супы, шоколад (суспензии); дым, мучная и сахарная пыль (аэрозоли). |
2. Ко́бальт — элемент побочной подгруппы восьмой группы четвёртого периода периодической системы химических элементов Д. И. Менделеева, атомный номер 27. Обозначается символом Co (лат. Cobaltum). Простое вещество кобальт (CAS-номер: 7440-48-4) — серебристо-белый, слегка желтоватый металл с розоватым или синеватым отливом. Существует в двух кристаллических модификациях: α-Co с гексагональной плотноупакованной решёткой, β-Co с кубической гранецентрированной решёткой , температура перехода α↔β 427 °C
Физические свойства
Кобальт — твердый металл, существующий в двух модификациях. При температурах от комнатной до 427 °C устойчива α-модификация. При температурах от 427 °C до температуры плавления (1494 °C) устойчива β-модификация кобальта (решётка кубическая гранецентрированная). Кобальт — ферромагнетик, точка Кюри 1121 °C. Желтоватый оттенок ему придает тонкий слой оксидов.
Химические свойства
Оксиды
На воздухе кобальт окисляется при температуре выше 300 °C.
Устойчивый при комнатной температуре оксид кобальта представляет собой сложный оксид Co3O4, имеющий структуру шпинели, в кристаллической структуре которого одна часть узлов занята ионами Co2+, а другая — ионами Co3+; разлагается с образованием CoO выше 900 °C.
При высоких температурах можно получить α-форму или β-форму оксида CoO.
Все оксиды кобальта восстанавливаются водородом:
![]()
Оксид кобальта (III) можно получить, прокаливая соединения кобальта (II), например:
![]()
Биологическая роль
Кобальт, один из микроэлементов, жизненно важных организму. Он входит в состав витамина В12 (кобаламин). Кобальт задействован при кроветворении, функциях нервной системы и печени, ферментативных реакциях. Потребность человека в кобальте 0,007-0,015 мг, ежедневно. В теле человека содержится 0,2 мг кобальта на каждый килограмм массы человека. При отсутствии кобальта развивается акобальтоз.
3. Химические свойства
Моносахариды вступают в химические реакции, свойственные карбонильной и гидроксильной группам. Характерная особенность моносахаридов — способность существовать в открытой (ациклической) и циклической формах и давать производные каждой из форм. Большинство моноз циклизуются в водном растворе с образованием гемиацеталей илигемикеталей (в зависимости от того, являются ли они альдозами или кетозами) между спиртом и карбонильной группой того же самого сахара. Глюкоза, например, легко образуетполуацетали, соединяя свои своим С1 и О5, чтобы сформировать 6-членное кольцо, названное пиранозид. Та же самая реакция может иметь место между С1 и О4, чтобы сформировать 5-членное фуранозид.
а) спиртовое брожение |
C6H12O6 |
б) молочно-кислое брожение |
C6H12O6 2CH3-CH(OH)-COOH молочная кислота |
в) масляно-кислое брожение |
C6H12O6 C3H7COOH + 2CO2 + 2H2O масляная кислота |
г) лимонно-кислое брожение |
C6H12O6 + O2 HOOC-CH2-C(OH)(COOH)-CH2-COOH + 2H2O лимонная кислота |
4. Карбо́новые кисло́ты — класс органических соединений, молекулы которых содержат одну или несколько функциональных карбоксильных групп -COOH.Кислые свойства объясняются тем, что данная группа может сравнительно легко отщеплять протон. За редкими исключениями карбоновые кислоты являются слабыми. Например, у уксусной кислоты CH3COOH константа кислотности равна 1,75·10−5. Ди- и трикарбоновые кислоты более сильные, чем монокарбоновые.
N 28
1. Высокомолекулярные соединения (полимеры), характеризуются молекулярной массой от нескольких тысяч до нескольких (иногда многих) миллионов. В состав молекул высокомолекулярных соединений (макромолекул)входят тысячи атомов, соединенных химическими связями. Любые атом или группа атомов, входящие в состав цепи полимераили олигомера, называются составным звеном. Наименьшее составное звено, повторением которого может быть описано строение регулярного (см. ниже) полимера, называется составным повторяющимся звеном. Составное звено, которое образуется из одной молекулы мономера при полимеризации, называется мономерным звеном (ранее иногда называлось элементарным звеном). Например, в полиэтилене [—СН2СН2—]n повторяющееся составное звено - СН2, мономерное -СН2СН2.
Название линейного полимера образуют прибавлением приставки "поли" (в случае неорганических полимеров - "катенан-поли"): а) к названию составного повторяющегося звена, заключенному в скобки (систематические названия); б) к названию мономера, из которого получен полимер (полусистематические названия, которые ИЮПАК рекомендует использовать для обозначения наиболее часто применяемых полимеров). Название составного повторяющегося звена образуют по правилам номенклатуры химической, например (первыми указаны полусистематические названия)
2.
N |
азот |
1s 22s 22p 3 |
P |
фосфор |
1s 22s 22p 63s 23p3 |
3. Глицерин.
Химические свойства
Химические свойства глицерина типичны для многоатомных спиртов.
Взаимодействие глицерина с галогеноводородами или галогенидами фосфора ведёт к образованию моно- и дигалогенгидринов.
Глицерин этерифицируется карбоновыми и минеральными кислотами с образованием соответствующих эфиров. Так, с азотной кислотой глицерин образует тринитрат — нитроглицерин (получен в 1847 г. Асканьо Собреро), использующийся в настоящее время в производстве бездымных порохов.
При дегидратации он образует токсичный акролеин:
HOCH2CH(OH)-CH2OH ![]() H2C=CH-CHO
+ 2 H2O,
H2C=CH-CHO
+ 2 H2O,
и окисляется до глицеринового альдегида СН2ОНСНОНСНО, дигидроксиацетона СН2ОНСОСН2ОН или глицериновой кислоты СН2ОНСНОНСООН.
Эфиры глицерина и высших карбоновых кислот — жиры являются важными метаболитами, важное биологическое значение играют такжефосфолипиды — смешанные глицериды фосфорной и карбоновых кислот.
4. Полисахариды
К полисахаридам относятся, в частности:
декстрин — полисахарид, продукт гидролиза крахмала;
крахмал — основной полисахарид, откладываемый как энергетический запас у растительных организмов;
гликоген — полисахарид, откладываемый как энергетический запас в клетках животных организмов, но встречается в малых количествах и в тканях растений;
целлюлоза — основной структурный полисахарид клеточных стенок растений;
хитин — основной структурный полисахарид экзоскелета насекомых и членистоногих, а также клеточных стенок грибов;
галактоманнаны — запасные полисахариды некоторых растений семейства бобовых, такие как гуаран и камедь рожкового дерева;
инулин - резервный углерод сложноцветных;
глюкоманнан — полисахарид, получаемый из клубней конняку, состоит из чередующихся звеньев глюкозы и маннозы, растворимое пищевое волокно, уменьшающее аппетит;
амилоид — применяется при производстве пергаментной бумаги;
многоглюкоза — многоконечный продукт гидролиза большинства многосахаридов.
N 29
1. ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ (полимеры), характеризуются мол. массой от неск. тысяч до неск. (иногда многих) миллионов. В составмолекул высокомолекулярных соединений (макромолекул)входят тысячи атомов, соединенных хим. связями. Любые атом или группа атомов, входящие в состав цепи полимера или олигомера, наз. составным звеном. Наим. составное звено, повторением к-рого м. б. описано строение регулярного (см. ниже) полимера, наз. составным повторяющимся звеном. Составное звено, к-рое образуется из одной молекулы мономера приполимеризации, наз. мономерным звеном (ранее иногда наз. элементарным звеном). Напр., в полиэтилене [—СН2СН2—]n повторяющееся составное звено - СН2, мономерное -СН2СН2.
Название линейного полимера образуют прибавлением приставки "поли" (в случае неорганич. полимеров -"катена-поли"): а) к названию составного повторяющегося звена, заключенному в скобки (систематич. названия); б) к названию мономера, из к-рого получен полимер (полусистематич. названия, к-рые ИЮПАК рекомендует использовать для обозначения наиб. часто применяемых полимеров). Название составного повторяющегося звена образуют по правилам номенклатуры химической. напр. (первыми указаны полусистематич. названия):
2. Комплексные соединения (лат. complexus — сочетание, обхват) или координационные соединения (лат. co — «вместе» и ordinatio — «упорядочение») — частицы (нейтральные молекулы или ионы), которые образуются в результате присоединения к данному иону (или атому), называемому комплексообразователем, нейтральных молекул или других ионов, называемых лигандами. Теория комплексных соединений (координационная теория) была предложена в 1893 г. А. Вернером.
Комплексные соединения мало диссоциируют в растворе (в отличие от двойных солей). Комплексные соединения могут содержать комплексный малодиссоциирующий анион ([Fe(CN)6]3−), комплексный катион ([Ag(NH3)2]+), либо вообще не диссоциировать на ионы (соединения типа неэлектролитов, например карбонилы металлов). Комплексные соединения разнообразны и многочисленны.
3. Восстанавливающие дисахариды
– это углеводы, которые при нагревании с водой в присутствии минеральных кислот или под влиянием ферментов подвергаются гидролизу, расщепляясь на две молекулы моносахаридов.
Наиболее широко распространенным дисахаридом является сахароза (тростниковый или свекловичный сахар). Получают его из сахарного тростника или из сахарной свеклы. В молоке содержится 5 % лактозы – молочного сахара. Мальтоза содержится в прорастающем зерне и образуется при гидролизе зернового крахмала. Целлобиоза является промежуточным продуктом при ферментативном гидролизе целлюлозы.
Строение. Молекула дисахарида состоит из двух молекул моносахаридов, соединенных гликозидной связью. В зависимости от того, какие атомы углерода участвуют в образовании гликозидной связи, молекула дисахарида может или не может содержать свободную карбонильную группу.
Дисахариды можно разделить на две группы: невосстанавливающие и восстанавливающие. Невосстанавливающие сахара не имеют ОН-группы ни при одном аномерном центре, восстанавливающие – имеют свободную ОН-группу при аномерном центре.
Невосстанавливающие сахара называют гликозил-гликозидами; восстанавливающие – гликозил-гликозами.
Мальтоза – восстанавливающий дисахарид, образующийся при ферментативном гидролизе крахмала. Мальтоза состоит из двух остатков D-глюкозы, соединенных гликозидной связью по положениям 1,4.
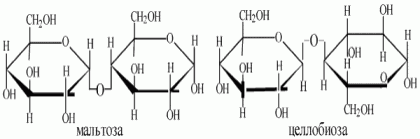
Сахароза состоит из остатков глюкозы и фруктозы, соединенных 1,2-гликозидной связью. У сахарозы полуацетальные гидроксильные группы обеих молекул моносахаридов участвуют в образовании гликозидной связи, вследствие чего сахароза является невосстанавливающим сахаром.
4. Реакции восстановления альдегидов и кетонов
Альдегиды при взаимодействии с водородом в присутствии Ni-катализатора образуют первичные спирты, кетоны - вторичные:
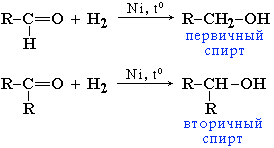
Восстановление. Альдегиды способны к восстановлению, основной продукт восстановления — первичные спирты.
![]()
Кетоны
Взаимодействие со спиртами:
CH3COCH3 + 2C2H5OH → C2H5—O—C(CH3)2—O—C2H5 + H2O
c реактивами Гриньяра:
C2H5—C(O)—C2H5 + C2H5MgI → (C2H5)3—COMgI → (C2H5)3—COH, третичный спирт. Реакции с альдегидами, и особенно с метаналем идут заметно активнее, при этом с альдегидами образуются вторичные спирты, а с метаналем — первичные.
Также кетоны реагируют с азотистыми основаниями, например, с аммиаком и первичными аминами с образованием иминов:
CH3—C(O)—CH3 + CH3NH2 → CH3—C(N—CH3)—CH3 + H2O
N 30
1. Буферные растворы (англ. buffer, от buff — смягчать удар) — растворы с определённой устойчивой концентрацией водородных ионов; смесь слабой кислоты и её соли (напр., СН3СООН и CH3COONa) или слабого основания и его соли (напр., NH3 и NH4CI). Величина рН буферного раствора мало изменяется при добавлении небольших количеств свободнойсильной кислоты или щёлочи, при разбавлении или концентрировании. Буферные растворы широко используют в различных химических исследованиях.
Буферные растворы имеют большое значение для протекания процессов в живых организмах. Например, в крови постоянство водородного показателя рН поддерживается буферными смесями, состоящими из карбонатов и фосфатов. Известно большое число буферных растворов (ацетатно-аммиачный буферный раствор, фосфатный буферный раствор, боратный буферный раствор, формиатный буферный раствор и др.)
Значение
pH буферного раствора можно рассчитать
по формуле: ![]() ,
где
,
где ![]() это
отрицательный десятичный логарифм от
константы диссоциации кислоты
это
отрицательный десятичный логарифм от
константы диссоциации кислоты ![]() .
.
По
сути ![]() .
.
3. Сахароза C12H22O11, или свекловичный сахар, тростниковый сахар, в быту просто сахар — дисахарид из группы олигосахаридов, состоящий из двух моносахаридов — α-глюкозы и β-фруктозы.
Сахароза является весьма распространённым в природе дисахаридом, она встречается во многих фруктах, плодах и ягодах. Особенно велико содержание сахарозы в сахарной свёкле и сахарном тростнике, которые и используются для промышленного производства пищевого сахара.
Сахароза имеет высокую растворимость. В химическом отношении сахароза довольно инертна, так как при перемещении из одного места в другое почти не вовлекается в метаболизм. Иногда сахароза откладывается в качестве запасного питательного вещества.
Сахароза, попадая в кишечник, быстро гидролизуется альфа-глюкозидазой тонкой кишки на глюкозу и фруктозу, которые затем всасываются в кровь. Ингибиторы альфа-глюкозидазы, такие, как акарбоза, тормозят расщепление и всасывание сахарозы, а также и других углеводов, гидролизуемых альфа-глюкозидазой, в частности, крахмала. Это используется в лечении сахарного диабета 2-го типа[1].
![]()
Трегалоза — углевод из группы невосстанавливающих дисахаридов. В природной трегалозе 2 остатка D-глюкозы связаны α,α-гликозидной связью.
Трегалоза впервые была выделена из спорыньи; содержится также в водорослях, дрожжах, высших грибах, лишайниках, в некоторых высших растениях, гемолимфе ряда червей и насекомых. Богатым источником трегалозы служит выделяющийся в результате укола насекомых экссудатясеня (Trehala manna). В туберкулёзных бациллах обнаружены производные трегалозы: высокотоксичный «корд-фактор» (трегалоза связана с 2 молекулами высшей жирной кислоты) и фосфоглюкан, в котором остатки трегалозы связаны фосфодиэфирной связью в линейную цепь. Биосинтез фосфата трегалозы происходит с участием уридиндифосфатглюкозы.
4. Иодное число — масса иода (в г), присоединяющегося к 100 г органического вещества.
Иодное число, которое характеризует содержание двойных связей в ненасыщенном соединении, определяют при исследовании жиров, а также при анализе жирных кислот и установлении содержания реагирующих с иодом примесей в ароматических углеводородах. Иодное число определяет общую ненасыщенность жиров. Чем выше иодное число, тем больше ненасыщенных кислот содержится в жире, то есть тем больше иода вещество может присоединить.