

Частичные агонисты Бупренорфин (бупренекс)
Бупренорфин - это полусинтетический опиоид, производное опиоида тебаина (рис. 8-26).
Фармакокинетика. Бупренорфин обладает высокой липофильностью [261]. В отношении этого препарата подтверждается ранее высказанное в этой главе положение, что кинетика обезболивающего действия анальгетика после его парентерального введения регулируется не общей фармакокинетикой, а преимущественно динамикой рецепторных диссоциаций [41, 44]. Диссоциация бупренорфина с μ-рецепторами происходит замедленно, и это объясняет продолжительность действия препарата, не совпадающую с временем его полувыведения из крови, составляющим 3-5 ч [262, 263]. Следовательно, прямой связи между содержанием бупренорфина в крови и его фармакологическим действием нет. Примерно 96% введенной дозы препарата связывается белками сыворотки [263].
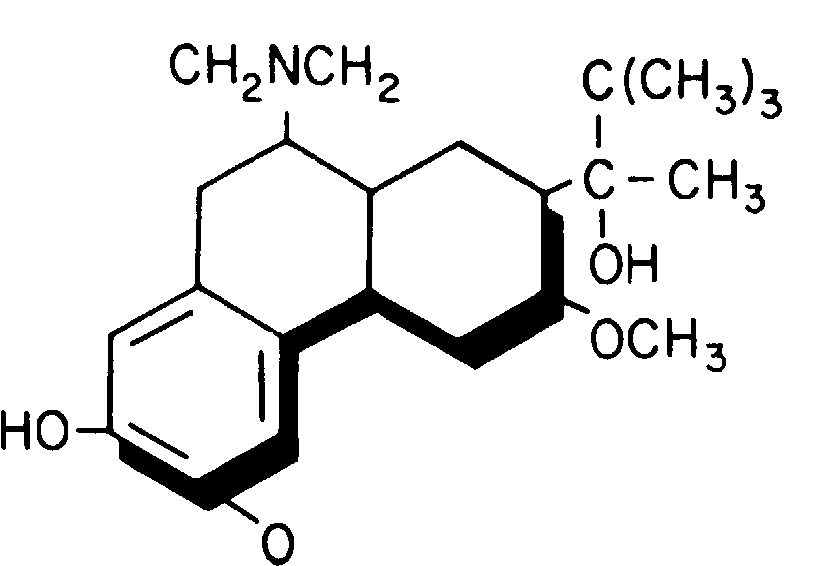
Рис. 8-26. Бупренорфин.
Фармакологическое действие. Бупренорфин - высокоактивный препарат.
При внутривенном его введении 0,3 мг этого препарата эквивалентны 10 мг морфина. На μ-рецепторы он действует как частичный агонист. Однако его аффинитет к этим рецепторам примерно в 50 раз сильнее, чем у морфина [264].
Побочные реакции на бупренорфин напоминают таковые у смешанных агонист-антагонистов. Седация и сонливость отмечаются почти у 50%, а тошнота и рвота - у 10-20% пациентов. Бупренорфин не вызывает психомиметических и дисфорических реакций, поскольку не является агонистом в отношении дельта-рецепторов.
Отмечено выраженное угнетение дыхания под влиянием бупренорфина. Это воздействие может быть весьма продолжительным из-за значительного и длительного аффинитета препарата к μ-рецепторам [265, 266]. По этой же причине реверсия подобного действия бупренорфина достигается лишь при назначении высоких доз налоксона [267]. Для поддержания адекватной вентиляции у больных, получивших большие дозы бупренорфина, можно применять доксапрам [268]. Обладая свойствами парциального агониста, бупренорфин способен несколько ослаблять действие высоких доз других μ-агонистов, уменьшая тем самым угнетение дыхания, вызванное этими опиоидами [269].
На сердечно-сосудистую систему бупренорфин оказывает такое же воздействие, как и морфин.
Использование в клинике и фармацевтические препараты. Бупренорфин обычно назначают внутримышечно или внутривенно в дозе 0,3 мг каждые 6 ч. При этом наиболее эффективно устраняются выраженные и тяжелые боли в послеоперационном периоде, болевой синдром при почечной колике, раке, инфаркте миокарда [270, 271].
В Европе бупренорфин выпускают для сублингвального употребления в дозах 0,4-0,8 мг. Этот путь введения обеспечивает хорошую послеоперационную аналгезию. Препарат также используется для АКП в послеоперационном периоде [272].
Для проведения ВВ-АКП бупренорфин теоретически является неподходящим препаратом. Аналгезия наступает медленно, но, развившись, продолжается длительно благодаря высокому аффинитету препарата к μ-рецепторам. По этой причине на фоне повторных назначений препарата может наступить передозировка, сопровождающаяся дальнейшим ухудшением дыхательной функции [273]. Однако были сообщения [274, 275] о том, что при проведении ВВ-АКТ бупренорфин обеспечивал хорошую аналгезию при минимальном угнетении дыхания.
Эпидуральное введение бупренорфина применяют для обезболивания после кесарева сечения и ортопедических операций [261, 276-278]. Бупренорфин, подобно налбуфину и буторфанолу, используют для устранения зуда после эпидурального введения μ-агонистов [279]. Субарахноидальному введению бупренорфина для аналгезии после операции кесарева сечения было посвящено всего одно исследование [280].
Бупренорфин выпускают в растворе концентрацией 0,3 мг/мл.
Дезоцин (далган)
Дезоцин относится к синтетическим препаратам морфинного ряда и обладает выраженными свойствами парциального μ-агониста (рис. 8-27).
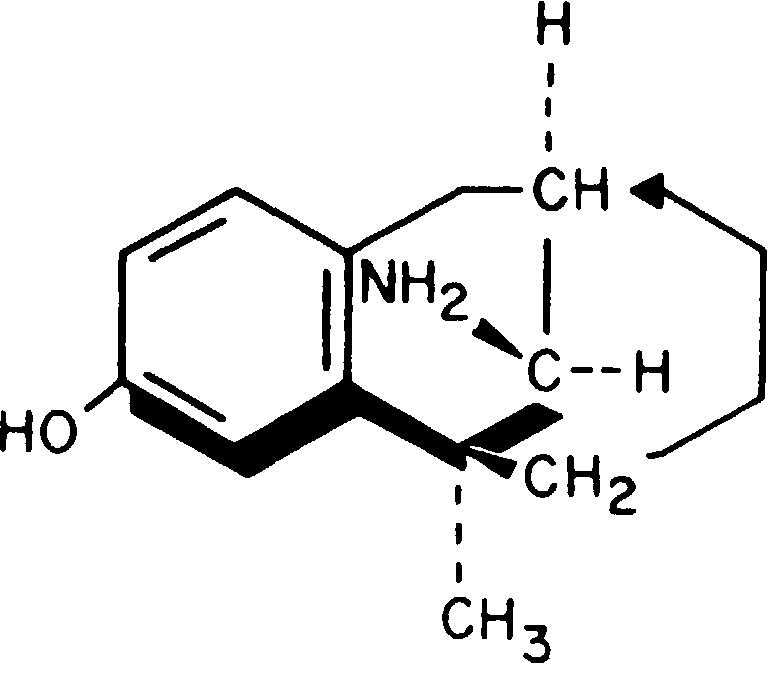
Рис. 8-27. Дезоцин.
Фармакокинетика. Опубликованы результаты всего одного исследования по фармакокинетике дезоцина у человека [280]. Препарат быстро распределяется в организме, но медленно выводится. Объем распределения у него весьма высок, что указывает на его интенсивное поглощение тканями. Дезоцин выделяется почками в неизмененном виде, а также подвергается метаболизму в печени [281].
Фармакологическое действие. По механизму действия дезоцин относится к частичным μ-агонистам, но одновременно он обладает свойствами дельта-агониста и в минимальной степени - капа-агониста. В отличие от налбуфина, пентазоцина и буторфанола дезоцин усиливает обезболивающий эффект даже при его назначении после других μ-агонистов [283]. При парентеральном введении действие дезоцина эквивалентно морфину. Аналгезический эффект дезоцина быстро ревертируется под влиянием налоксона как в эксперименте, так и в клинике [284, 285]. Препарат не так плотно фиксируется μ-рецепторами, как бупренорфин.
Дезоцин вызывает побочные реакции, характерные для агонист-антагонистов в целом: седацию (иногда продолжительную), тошноту и рвоту. Седативный эффект наступает через 1 ч после внутримышечного введения 10-15 мг дезоцина [286]. Опубликовано сообщение о дисфорических реакциях после введения больших доз дезоцина, хотя аффинитет препарата к дельта-рецепторам невелик [287].
Угнетение дыхания под влиянием повышенных доз дезоцина имеет характер «потолка» действия [283, 284]. При дозах свыше 0,3 мг/кг не происходит дальнейшего увеличения ни аналгезии, ни угнетения дыхания [288].
Влияние дезоцина на сердечно-сосудистую систему пока недостаточно изучено. По данным единичных исследований больных, подвергшихся катетеризации сердца, дезоцин в дозе 0,125 мг/кг вызывает непродолжительное повышение давления в легочной артерии и сосудистого сопротивления в малом круге. Частота сердечных сокращений и артериальное давление не изменяются [288, 289].
Вероятность развития наркотической зависимости минимальная, хотя имеются сообщения о наркотическом потенциале дезоцина [290, 291]. Возможности дезоцина провоцировать развитие синдрома отмены у пациентов с зависимостью от опиоидов не изучены.
Использование в клинике и фармацевтические препараты. Дезоцина лактат выпускают в растворах концентрацией 5, 10 и 15 мг/мл. Было проведено несколько клинических исследований по парентеральному применению дезоцина с целью послеоперационной аналгезии [292-295], однако истинная его роль в этом отношении остается неясной. Отсутствует также опыт по использованию этого препарата для АКП или для энидуральной аналгезии.
АНТАГОНИСТЫ
Небольшие изменения химической структуры μ-агонистов способны трансформировать их в антагонисты по отношению к одному или многим рецепторам [75]. Налоксон является N-аллил-(—CH2—CH=CH2) производным оксиморфона (см. рис. 8-15). Налтрекс имеет циклопропилметиловую группу (—СН2—<) на третьем атоме азота. Известны и другие опиоидные антагонисты, и первый из них - налорфин, а также налмефен и холецистокинин [296-298]. Они не обсуждаются в данной главе.
Налоксон и налтрексон являются чистыми антагонистами в отношении μ-, капа- и дельта-рецепторов. Оба последних препарата обладают высоким аффинитетом к μ-рецепторам. Их аффинитет к дельта- и капа-рецепторам менее выражен, но тем не менее они вытесняют агонисты из связи с соответствующими рецепторами. Однако после вытеснения налоксон и налтрексон не активируют опиоидные рецепторы, вызывая антагонистическое действие.
Парентеральные антагонисты
Налоксон (наркан)
Фармакокинетика. Внутривенное введение налоксона в дозе 1-4 мкг/кг приводит к реверсии аналгезии и дыхательной депрессии, вызванных опиоидами. Продолжительность этого действия невелика (30-45 мин), что, вероятно, связано с метаболизмом и быстрым вымыванием препарата из рецепторов в мозге [299]. Поэтому для поддержания антагонистического действия препарата бывают необходимы повторные его введения.
Налоксон подвергается метаболизму при первом прохождении в печени (рис. 8-28). Время его полувыведения составляет 64 мин [300]. Особенно интенсивный метаболизм наблюдается при энтеральном приеме налоксона.
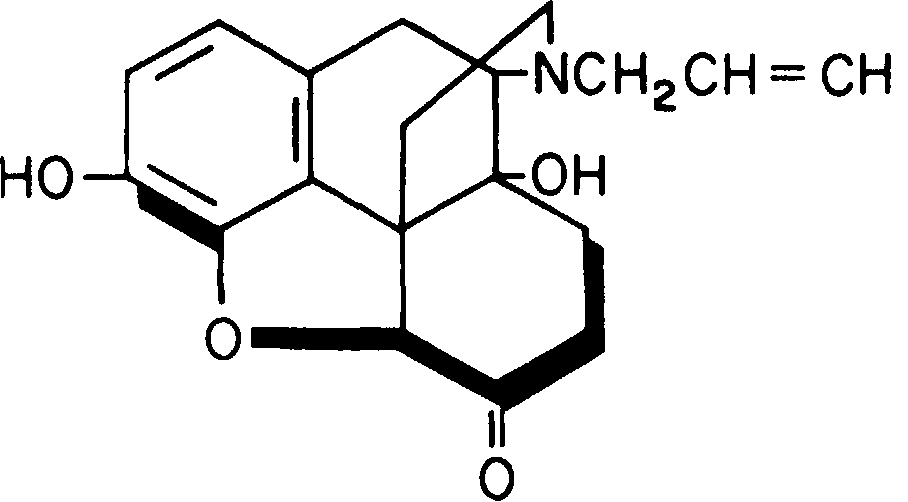
Рис. 8-28. Налоксон.
Фармакологическое действие. В отношении опиоидов, назначаемых обычными путями (не нейрогенными), налоксон действует как антагонист, устраняя вызванное ими нарушение дыхания и аналгезию (см. табл. 8-6). При специальном подборе дозировок можно сохранить, хотя бы частично, аналгезию при минимально выраженной дыхательной депрессии. Однако тошнота, рвота и стимуляция сердечно-сосудистой деятельности могут сопровождать частичное ослабление аналгезии.
Действие налоксона, введенного эпидурально, возрастает соответственно повышению дозировки. При инфузии налоксона в дозе 5 мкг/кг в 1 ч качество вызываемой эпидуральным введением морфина аналгезии не изменяется, но устраняются нарушения дыхания. Инфузия налоксона в дозе 10 мкг/кг в 1 ч уже снижает аналгезию [301]. Введение налоксона в дозах 5-10 мкг/кг в 1 ч устраняет как депрессию дыхания, так и аналгезию, вызванную фентанилом [302]. Причины столь выраженной разницы между морфином и фентанилом неясны.
Появление тошноты и рвоты непосредственно связано со скоростью введения налоксона [303, 304]. Малые дозы препарата, назначаемые каждые 2-3 мин, снижают частоту этих побочных реакций. Налоксон может увеличивать нагрузку на сердечно-сосудистую систему. Это выражается активацией симпатической нервной системы: тахикардия, гипертензия, аритмия, отек легких [305-307].
Использование в клинике и фармацевтические препараты. Больным с выраженной седацией и угнетением дыхания (менее 8 вдохов в 1 мин) налоксон назначают внутривенно в начальной дозе 10 мкг. Эта доза удваивается каждые 23 мин (20, 40, 80, 160 мкг) вплоть до пробуждения пациента и нормализации дыхания. В последующем необходимо продолжать инфузии налоксона или вводить повторно те же дозы. Пациентам в состоянии апноэ и не пробуждающимся налоксон вводят в дозе 0,4 мг. Налоксона гидрохлорид выпускают в растворе концентрацией 0,02, 0,4 и 1 мг/мл. Для взрослых чаще всего используют ампулы с содержанием налоксона 0,4 мг/мл.