

Синтетические опиоиды Леворфанол (лево-дроморан)
Леворфанол единственный доступный μ-агонист из группы морфина (см. рис. 8-3; рис. 8-16). Правовращающий изомер (декстрометорфан) обладает такой же противокашлевой активностью, как кодеин, но не оказывает обезболивающего действия и не вызывает наркомании.
Фармакологический профиль леворфанола такой же, как у морфина, он подвержен значительному метаболизму при первом прохождении, хотя возможны индивидуальные отклонения [149, 150]. Соотношение активности при оральном и парентеральном назначении такое же, как у кодеина и оксикодона. Леворфанол в 7 раз активнее морфина при энтеральном и в 5 раз при парентеральном введении (см. табл. 8-8).
Фармакокинетика. Леворфанол быстро рассасывается после подкожного введения. Максимальная аналгезия наступает через 60-90 мин, а продолжительность обезболивания такая же, как после парентерального введения морфина. Метаболизм препарата осуществляется медленно (время полувыведепия 11 ч). Поэтому повторные инъекции препарата через короткие интервалы времени могут вызвать его аккумуляцию [150].
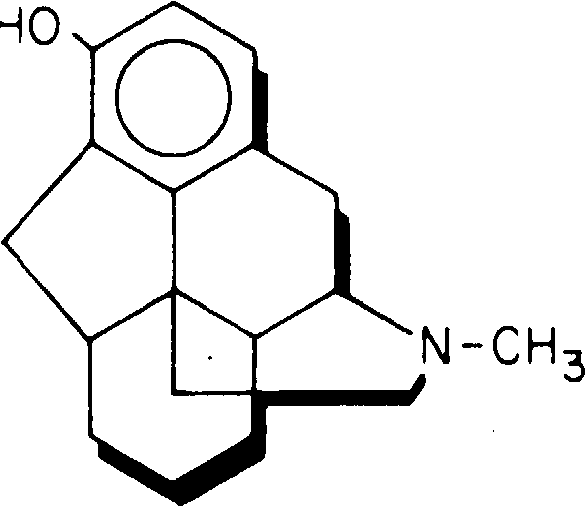
Рис. 8-17. Меперидин.
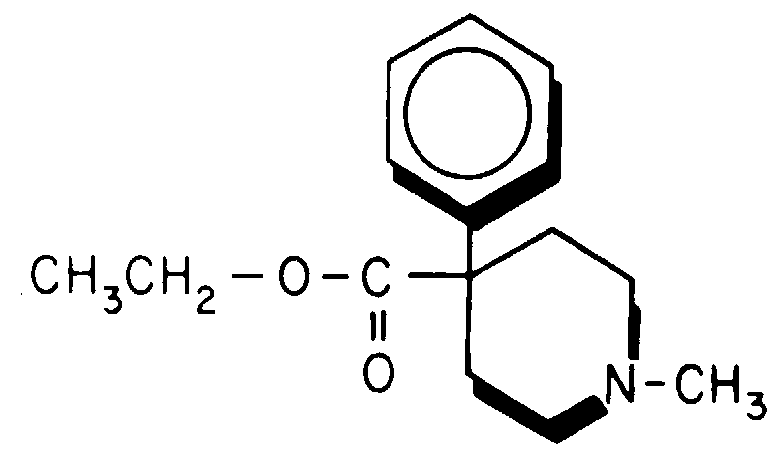
Рис. 8-16. Леворфанол. Обезболивающими свойствами обладает только его левовращающий изомер.
Использование в клинике и фармацевтические препараты. Леворфанол чаще всего используют при хронических болях, преимущественно при ведении больных раком [52, 151].
Выпускают препарат в виде леворфанола гартарата, в таблетках по 2 мг или в растворе для инъекций с концентрацией вещества 2 мг/мл.
Меперидин (демерол)
Меперидин - один из препаратов группы фенилпиперидина (рис. 8-17) относится к опиоидным μ-агонистам. Преимущественно используемые опиоидные анестетики фентанил, суфенганил и алфентанил (рис. 8-18, 8-19 и 8-20) являются аналогами меперидина.
Фармакокинетика. При энтеральном назначении препарат подвергается метаболизму в печени, и биологическое действие оказывает 45-75% введенной дозы. Меперидин всасывается медленнее, пик его концентрации в крови наступает через 2 ч после приема внутрь [152].
Скорость рассасывания препарата при внутримышечном введении весьма вариабельна, поэтому обезболивающее его действие неустойчивое и часто недостаточное [54, 153].
После внутривенного введения меперидин переходит из крови в ткани, распределение его завершается через 30-45 мин, что гораздо медленнее, чем у морфина (10 мин после внутривенного введения).
Время полувыведения меперидина составляет 3-4,4 ч [154]. Около 60% препарата связывается белками плазмы. Ослабление связывания препарата белками крови у пожилых лиц может привести к увеличению содержания свободной фракции меперидина и вызвать повышенную чувствительность к нему [155].
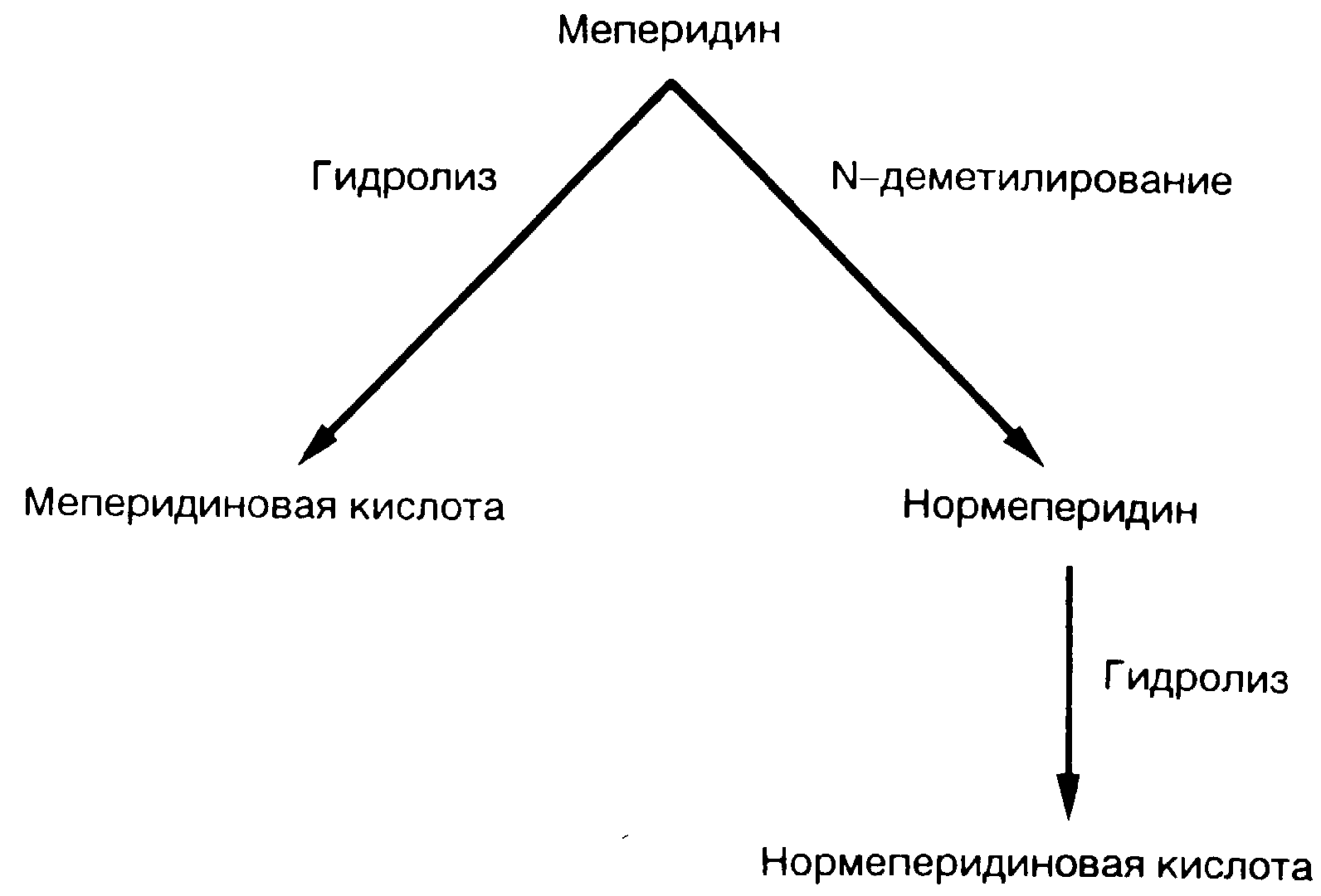
Рис. 8-21. Метаболизм нормеперидина в печени.
Метаболизм: значение метаболита нормеперидина. Меперидин интенсивно разрушается в печени (рис. 8-21). Примерно 90% всей введенной дозы подвергается N-деметилированию с образованием нормеперидина и гидролизу до меперидиновой кислоты [156, 157]. С мочой выделяется менее 5% введенной дозы препарата. Нормеперидин также подвержен гидролизу с образованием нормеперидиновой кислоты. Кислотные метаболиты не обладают биологической активностью и выделяются с мочой в неизмененном и частично в конъюгированном виде [158].
Выделение меперидина с мочой зависит от показателя рН. Если рН мочи опускается ниже 5,0, то с ней выделяется в неизмененном виде около 25% принятой дозы опиоида. Скорость выведения меперидина можно увеличить, способствуя подкислению мочи [159].
Время удаления меперидина колеблется от 15 до 40 ч, препарат можно обнаружить в моче даже через 3 дня после его приема. Нормеперидин оказывает возбуждающее действие на ЦНС, его токсические эффекты выражаются миоклонусом и судорогами [157, 160, 161]. Поэтому назначение меперидина больным с почечной недостаточностью может привести к его накоплению и развитию нормеперидиновой интоксикации [160, 162]. Цирроз печени иногда становится причиной сниженного клиренса и длительной задержки нормеперидина в крови. В то же время больные с циррозом печени до некоторой степени защищены от интоксикации нормеперидином вследствие снижения метаболизма препарата. При повторных назначениях препарата опасность токсических реакций возрастает [163].
Фармакологическое действие. Меперидин почти в 10 раз слабее морфина при энтеральном приеме и в 7-10 раз - при парентеральном введении. В аналгезирующих дозах не оказывает видимого влияния на сердечнососудистую систему. Меперидин в противоположность морфину и другим опиоидам не замедляет ритма сердца. Напротив, благодаря своему структурному сходству с атропином (см. рис. 8-10) он способен спровоцировать тахикардию. В больших дозах меперидин снижает сократительную способность миокарда, величину ударного объема и одновременно повышает давление наполнения. Отрицательный инотропный эффект меперидина проявляется при его назначении в дозе 2-2,5 мг/кг [89, 90].
Незначительное антиспастическое действие этого препарата было отмечено уже при первом описании свойств этого вещества Eisleb и Shaumann в 1939 г. При назначении в эквивалентных с морфином аналгезических дозах не происходит столь значительного спазма желчных путей [164]. Слабое влияние на гладкую мускулатуру делает меперидин препаратом выбора среди других опиоидов при лечении больных с почечной коликой.
В отличие от других опиоидов меперидин чаще вызывает мидриаз, а не миоз, что отражает его атропиноподобные свойства.
Использование в клинике и фармацевтические препараты. Меперидин назначают внутримышечно при сильных болях по 75-100 мг [165, 166]. Повторные введения препарата могут потребоваться каждые 2-4 ч, так как продолжительность создаваемой им аналгезии меньше, чем у морфина. При ведении послеоперационных больных меперидин назначают в инфузиях, нагрузочная доза составляет 0,5-1,5 мг/кг. Спустя 30-60 мин переходят на поддерживающие дозы 0,25/0,75 мг/мин [167]. Поддерживающие дозы необходимо часто корригировать (см. гл. 10). Меперидин чаще других препаратов используют для ВВ-АКП [138, 168, 169].
Меперидина гидрохлорид выпускают в таблетках по 50 и 100 мг, а также в растворах (50 мг в чайной ложке). Препарат для парентерального применения выпускается в различных концентрациях.
Фентанил
Фентанил является производным меперидина и входит в группу фенил-пиперидина (см. рис. 8-18). Обезболивающее действие препарата в 75-125 раз сильнее, чем у морфина [170].
Фармакокинетика. Более высокая, чем у морфина, растворимость в липидах объясняет быстрое наступление эффекта после введения фентанила (в течение 30 с) и небольшую продолжительность действия. Высокая липофильность объясняет быстрое и значительное распределение препарата в тканях. В хорошо перфузируемых тканях высокая концентрация фентанила достигается быстро. Эффект от действия фентанила вскоре прекращается в связи с быстрым высвобождением его из жировой ткани и из скелетных мышц и соответственно снижением его содержания в плазме крови [171].
Таким образом, кратковременность действия однократной дозы фентанила отражает быстрое его потребление тканями и столь же быстрое высвобождение с падением уровня препарата в крови. При повторных введениях или при непрерывной инфузии фентанила может наступить насыщение им неактивных жировых и мышечных депо. В этом случае темпы снижения его концентрации в крови замедляются и действие фентанила удлиняется. Следовательно, снижение уровня препарата в крови отражает его удаление, а не распределение в тканях [172].
Метаболизм фентанила проходит путем деалкилирования, гидроксилирования и амидного гидролиза с образованием норфентанила и деспропионилнорфентанила, которые выводятся с мочой и желчью. В неизмененном виде с мочой выделяется не более 8% принятой дозы препарата. В противоположность меперидину норметаболиты фентанила неактивны и не оказывают стимулирующего влияния на ЦНС. Считается, что фентанил является препаратом выбора для больных с нарушением функции почек [173, 174].
Несмотря на кратковременность действия фентанила, выведение его из организма происходит относительно медленно. Время полувыведения составляет 185-219 мин, что отражает большой объем распределения препарата в тканях (см. табл. 8-7). Последнее обстоятельство связано с высокой растворимостью препарата в липидах. Цирроз печени не оказывает заметного влияния на сроки выведения фентанила [175]. У лиц пожилого возраста удаление препарата замедлено еще больше, что связано с более медленным клиренсом. Объем распределения препарата у лиц пожилого возраста остается таким же, как и у молодых [176, 177]. Следовательно, в пожилом возрасте действие фентанила может пролонгироваться.
Фармакологическое действие. Фентанил влияет на ЦНС как депрессант, вызывая аналгезию и подавляя дыхание. Весьма примечательно, что в отличие от меперидина фентанил в малых дозах (1-2 мкг/кг) обладает слабым гипнотическим и седативным действием (см. табл. 8-9). Большие дозы, не применяемые в практике обезболивания (50-150 мкг/кг), вызывают глубокую седацию вплоть до потери сознания.
Фентанил при парентеральном введении почти в 100 раз активнее морфина, но, несмотря на это, при введении названных средств в эквивалентных дозах угнетение дыхания развивается в одинаковой степени.
В отличие от морфина фентанил даже в больших дозах не высвобождает гистамин [95]. Введение фентанила способно индуцировать брадикардию, но выраженной степени она достигает только при анестезирующих дозах препарата [178].
Использование в клинике и фармацевтические препараты. Фентанила цитрат применяют в концентрации 50 мкг/мл. Препарат выпускают в таблетках вместе с дроперидолом (инновар), однако эта комбинация малопригодна для устранения послеоперационной боли.
Продолжаются интенсивные исследования по внутривенному (см. гл. 10), эпидуральному и субарахноидальному введению фентанила (гл. 11 и 12). Сведений о фармакокинетике фентанила после его внутримышечного введения в литературе нет.
В недавнем прошлом фентанил не назначали энтерально из-за выраженного метаболизма в печени и слабой усвояемости в биологически активной форме (32%) [79]. Однако созданы новые модификации препарата, например, в виде трансмукозального фентанила цитрата для энтерального употребления, и его усвояемость повышается до 52% [179]. Препарат обычно применяют для предоперационной седации в педиатрии [180, 181] и для купирования болей у больных раком [182, 183]. Дозы варьируют от 10 до 25 мкг/кг. Побочное действие при таких дозах проявляется легким зудом в области лица (65-85%) и слабым зудом всего тела (10-30%) либо выраженной тошнотой (30-37%) [183-186]. Трансмукозальный фентанила цитрат не используют для преодоления послеоперационных болей. Чаще его применяют как дополнительное средство для обезболивания и седации в палатах интенсивной терапии [186]. Однако в последнее время описаны показания для интраназального применения фентанила с целью снятия болей в послеоперационном периоде [187].
Трансдермальное введение фентанила. Разработка трансдермальных методов введения (транстермальные терапевтические системы-ТТС) эстрогенов, клофелина и скополамина вызвало интерес к подобному же методу назначения липофильных опиоидов [188]. Липофильность фентанила, возможность седативного действия и влияние на сердечно-сосудистую систему - все это делает весьма привлекательным трансдермальный путь его введения.
Разработаны четыре системы для эффективного трансдермального введения препаратов. В отношении фентанила используют ТТС, основанную на принципе мембранного проникновения (рис. 8-22). Фентанил помещают в мелкий резервуар из непроницаемой пленки, верхнюю поверхность которого прикрывают мелкопористой мембраной, ограничивающей скорость трансдермального проникновения. Плотный контакт с кожей обеспечивается специальным адгезивным полимером, нанесенным на внешнюю сторону микропористой мембраны. В резервуаре содержится небольшое количество фентанила (до 10 мг) в гелевом матриксе, который и определяет скорость диффузии. При необходимости повысить дозировку достаточно увеличить поверхность контактирующей с кожей ТТС так, чтобы поддерживалось постоянное поступление фентанила со скоростью 25, 50, 75 или 100 мкг/ч в течение до 3 дней [188].
Основным препятствием для поступления препарата является роговой слой эпидермиса, поскольку диффузия происходит в основном с участием внутриклеточных липидных сред [190]. Кожа представляет собой как бы резервуар, который должен наполниться, прежде чем будет поддерживаться постоянная абсорбция. В последующем поступление фентанила будет продолжаться даже после удаления ТТС [188].
После наложения ТТС концентрация фентанила в плазме крови возрастает на протяжении 12-18 ч, пока не стабилизируется на определенном уровне (плато). Это состояние соответствует окончательному формированию депо препарата в коже. Концентрация фентанила в плазме крови остается постоянной весь срок прикрепления ТТС к коже. После удаления ТТС уровень препарата в крови постепенно снижается. Периодполу выведения составляет 15-21 ч, что соответствует опорожнению фентанилового депо [190-192].
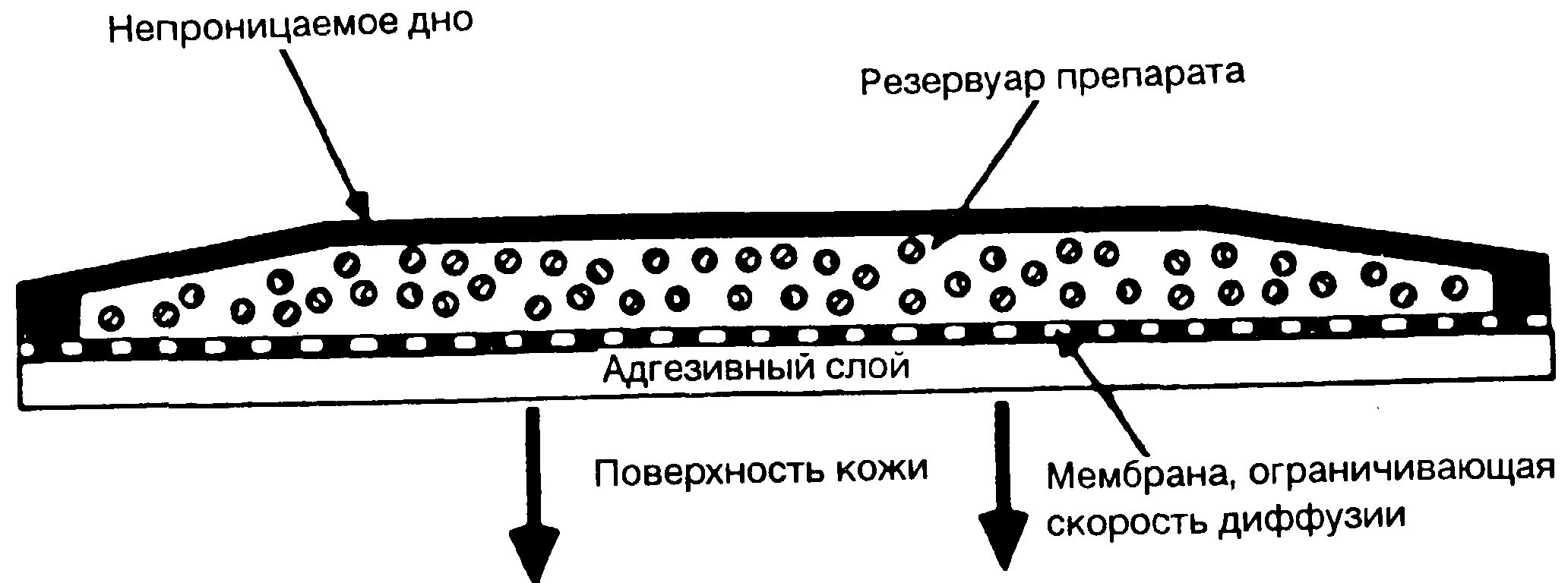
Рис 8-22 Мембранная пермеация (проникновение) при трансдермальном терапевтическом введении опиоидов. (Из Tarver и Stanley [189], с разрешения.)
Таблица 8-12. Преимущества и недостатки фентанил-ТТС при ведении больных с послеоперационными болями
Преимущества |
Недостатки |
Снижение метаболизма первого прохождения
Стабильная концентрация в крови Простота применения Доза рассчитана на несколько дней Не нужны иглы и инъекции Эффективная техника аналгезии |
Невозможность титрования дозы в соответствии с повышением или снижением потребности в аналгсзии Заранее выбранная доза Медленное наступление действия Необходимость дополнительной аналгезии Остаточное депо после удаления ТТС Потенциальная наркотическая зависимость |
По данным всех исследований, посвященных послеоперационной боли, при использовании фентанил-ТТС потребность в опиоидах снижается. Побочные реакции проявляются тошнотой и рвотой (30-85%) или угнетением дыхания [191-196].
Преимущества и недостатки фентанил-ТТС приведены в табл. 8-12. Основной недостаток этой методики связан с титрованием дозы препарата. Эффективное преодоление острой боли зависит именно от возможности варьировать дозировку в соответствии со степенью выраженности болевых ощущений. К сожалению, фентанил-ТСС имеет такие же недостатки, как и другие системы фиксированного дозирования, делающие их малоприемлемыми для преодоления послеоперационной боли (в нашем понимании этой проблемы). Наиболее целесообразно применять фентанил-ТТС для снятия болей у онкологических больных. Трансдермальное назначение фентанила может стать эффективным промежуточным методом на этапе перехода от орального к парентеральному введению опиоидов.
Суфентанил
Суфентанил (см. рис. 8-19) является тиаминовым аналогом фентанила и входит в группу фенилпиперидиповых синтетических опиоидов. Он активнее фентанила в 5-10 раз, соответственно выше и аффинитет его рецепторного связывания.
Фармакокинетика. Высокая растворимость суфентанила в липидах (разделительный коэффициент 1250) согласуется с быстрым проникновением препарата через гематоэнцефалический барьер и объясняет быстрое наступление его действия. Высокий тканевый аффинитет (обусловленный липофильностью препарата) приводит к быстрому его распределению в организме. Как и у фентанила, быстрое перераспределение суфентанила в неактивные ткани (жир, скелетные мышцы) резко ограничивает его действие, особенно при назначении небольших доз [198].
Суфентанил подвержен быстрому метаболизму путем N-деалкилирования пиперидинового азота и 0-деметилирования [199]. Продукты деалкилирования не обладают биологической активностью. Десметилсуфентанил, образующийся при деметилировании, сохраняет примерно 10% активности суфентанила. С мочой выводится менее 1% неизмененного суфентанила.
Метаболиты суфентанила выделяются как с мочой, так и с калом. Около 30% выделяющихся метаболитов конъюгируют, преимущественно это происходит с десметилсуфентанилом. Вследствие значительной способности к конъюгации и продукции активных метаболитов назначать препараты пациентам с почечными заболеваниями следует с осторожностью [200, 201].
Объем распределения суфентанила несколько меньше, а скорость выделения вдвое меньше, чем у фептанила. Суфентанил интенсивно связывается с белками плазмы (90% препарата). Клиренс препарата ослабевает у лиц пожилого возраста, однако время полувыведения не изменяется из-за уменьшения объема перераспределения. Тем не менее у пожилых пациентов может наблюдаться и пролонгированное действие препарата [202].
Фармакологическое действие. Клинико-фармакологический профиль суфентанила почти такой же. как у фентанила, но более выражено седативное действие. При назначении суфентанила несколько чаще развиваются брадикардия, миоз, yгнетение дыхания, тошнота, рвота и спазм гладких мышц.
Применение в клинике и фармацевтические препараты. Для инъекций выпускают суфентапил цитрат в концентрации 50 мкг/мл. Для энтерального приема препарат не производят, продолжается его изучение с целью трапсдсрмального назначения [203, 204].
Опыт применения суфентанила для купирования послеоперационной боли весьма ограничен. Основные исследования проводятся по применению суфентанила при анестезии, контролируемой пациентом (АКН), и при эпидуральной анестезии [205 208].
Алфентанил
Алфентанил (см. рис. 8-20) является производным фептанила. Он слабее последнего в 5-10 раз, а продолжительность его действия на 1/3 короче, чем у фентанила.
Фармакокинетика. Главными фармакологическими особенностями алфентанила, объясняющими его действие в клинике, служат его низкий рН (6,5) и малый объем распределения [209-211]. Аналгезия после внутривенного введения алфентанила наступает очень быстро (через 1-2 мин). Это может быть связано с низкими величинами рКа, поскольку почти 90% препарата ионизируется при рН 7,4 (см. табл. 8-7). Благодаря высокой ионизации препарат быстро проникает через гематоэнцефалический барьер несмотря на слабую растворимость в липидах.
Объем распределения алфентапила в 4-5 раз меньше, чем у фентанила [209, 210]. Слабый тканевый аффинитет алфентанила отражает его слабую растворимость в липидах и высокую степень связывания с протеинами.
Небольшая продолжительность действия алфентанила обусловлена его перераспределением в неактивные ткани и столь же быстрым метаболизмом в печени, как и у суфентанила. С мочой выделяется менее 1% неизмененного алфентанила.
Время полувыведения препарата составляет 70-98 мин [210]. У больных с циррозом печени этот показатель возрастает до 219 мин [211]. Кроме того, возрастание свободной фракции алфентанила у больных с печеночной патологией объясняется изменением состава белков крови и нарушением их способности связывать препарат. Более медленное выделение алфентанила и увеличение его свободной фракции у больных с циррозом печени могут привести к усилению и к удлинению действия препарата. У больных с патологией почек клиренс алфентанила не нарушается, но объем распределения может измениться в зависимости от связывания препарата белками плазмы. Таким образом, выделение алфентанила не нарушается при патологии почек, но нарушение связывания с белками способно влиять на его распределение.
Использование в клинике и фармацевтические препараты. Подобно суфентанилу, лучше всего изучено применение алфентанила для эпидуральной анестезии [213, 214] и для внутривенной анестезии, контролируемой пациентом (АКП) [213-216].
Краткость действия алфентанила имеет значение для его использования при АКП. В то же время краткосрочность действия может потребовать чрезвычайно больших дозировок и приведет к истощению собственных возможностей пациента в процессе АКП [216]. К сожалению, проведение инфузий, ориентированных на нижнюю границу потребностей пациента, не обеспечивает достаточного обезболивания [215, 216].
В настоящее время алфентанил выпускают только для инъекций, концентрация раствора 500 мкг/мл.