

1. Реакция гидрирования
Реакции гидрирования алкенов являются иллюстрацией ненасыщенного характера алкенов в силу отмеченной выше слабости -связи. Последняя легко рвется под действием гидрирующего агента и по освобождающимся валентностям присоединяются атомы водорода.
![]()
В качестве катализаторов используют Pd, Pt, Ni, нанесенные на пористые носители: -AI2O3, SiO2, алюмосиликаты, активированный уголь.
Присоединение водорода происходит в цис-положение. Такой характер присоединения обусловлен геометрией активированного комплекса реакции гидрирования на поверхности катализатора:
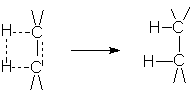
Экспериментальное подтверждение этого является образование цис-1,2-диметилциклогексана при гидрировании 1,2-диметилциклогексена
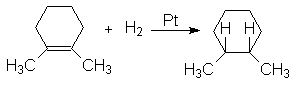
Гидрирование на металлических катализаторов часто осложняется изомеризацией алкенов и разрывом простых углерод-углеродных связей.
Более удобными в этом отношении являются катализаторы гомогенного гидрирования – комплексы хлоридов родия или рутения с трифенилфосфином (C6H5)3P (катализаторы Уилкинcона), например, хлорид трис-(трифенилфосфин)родия, получаемый при взаимодействии хлорида родия (III) с трифенилфосфином.
![]() Легкость
гидрирования в этих реакциях зависит
от степени замещения при двойной связи:
чем больше степень замещения, тем
медленнее гидририруется двойная связь.
Это открывает возможность селективного
гидрирования соединений, содержащих
различные по степени замещенности
двойные связи:
Легкость
гидрирования в этих реакциях зависит
от степени замещения при двойной связи:
чем больше степень замещения, тем
медленнее гидририруется двойная связь.
Это открывает возможность селективного
гидрирования соединений, содержащих
различные по степени замещенности
двойные связи:
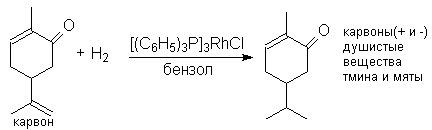
Можно видеть, что замещенная двойная связь и карбонильная группа не подвергаются гидированию.
Показано, что гомогенное гидрирование в присутствии указанных катализаторов происходит в цис-положение.
Недостаток катализаторов Уилкинсона – невозможность его использования для гидрирования соединений, содержащих альдегидную группу из-за осложнений основной реакции декарбонилированием
RCHO RH + CO
2. Реакции электрофильного присоединения по двойной связи алкенов.
а) Реакции галогенирования.
Алкены легко реагируют в обычных условиях с растворами CI2 или Br2 в инертном растворителе (например, CCI4) согласно стехиометрическому уравнению:
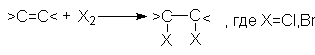
J2 в этих реакциях значительно менее активен.
Механизм реакций галогенирования может быть представлен последовательностью стадий (1) – (5), предполагающей образование -комплексов между молекулами галогена и кратной связью алкена с последующим его переходом в -комплекс (I) и (или) неклассический катион (II). Заключительными стадиями являются реакции (4) и (5), представляющие собой быструю нуклеофильную атаку интермедиатов I и II с образованием продукта галогенирования.
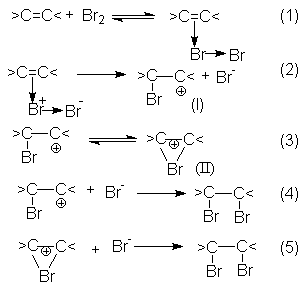
Скорость–определяющей стадией в этих реакциях является переход - комплекса в -комплекс.
Аргументами в пользу этого являются:
кинетическое уравнение реакций галогенирования двойных связей
r = k[>C=C<][Br2]
ускорение реакций сильно сольватирующими растворителями
ряд реакционной способности алкенов в реакциях галогенирования:
(CH3)2C=CH2 > CH3-CH=CH2 > CH2=CH2
соответствующий ряду стабильности соответствующих -комплексов:
![]()
В общем случае повышение реакционной способности алкенов обусловлено наличием при углеродных атомах кратных связей электронодонорных заместителей.
В то же время образование продуктов транс-присоединения, наблюдаемое при галогенировании циклоалкенов, может быть обосновано исключительным образованием неклассических циклических ионов (II), в которых атом галогена блокирует подход галогенид-аниона со стороны занятого им положения.
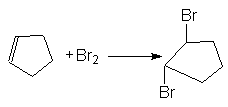
В этой связи следует предположить, что время жизни классических карбкатионов в таких реакциях слишком мало и они быстро изомеризуются в циклический катион по реакции (3).
В то же время если заместители при атомах углерода двойной связи существенно отличаются по своей электронодонорной способности, то образование открытого карбкатиона может стать энергетически более выгодным и реакция присоединения должна идти нестереоспецифично, поскольку открытый карбкатион равновероятно атакуется с противоположных сторон галогенид анионом. Такая ситуация реализуется при бромировании п-метоксипропенилбензола
СH3-O-C6H4CH=CH-CH3 + Br2 CH3O-C6H4CHBr-CHBr-CH3
когда образуется смесь энантиомеров.
Другим тестом на протекание реакции через классических катион является наличие побочной реакции полимеризации.
Таким образом, в зависимости от строения алкена реакция может идти как через классический, так и через мостиковый ион, причем симметрия последнего зависит от природы заместителей при двойной связи, и связь С…Hal в мостиковом катионе в меньшей степени связана с атомом, при котором существуют структурные возможности для делокализации положительного заряда. На заключительной стадии именно с этим атомом взаимодействует нуклеофил.
Ассимметриченое строение алкенов обусловливает характер образующихся продуктов в реакциях сопряженного присоединения, когда при проведении реакции галогенирования в реакционной массе присутствуют посторонние нуклеофильные агенты, например, другие галогениданионы, вода, спирты и т.д.
CH3CH=CH2 + Br2 + Cl- CH3CHCl-CH2Br + Br- или
CH3CH=CH2 + Br2 + NaCl CH3CHCl-CH2Br + NaBr
CH3CH=CH2 + Cl2 + H2O CH3CHOH-CH2Cl + HCl
Последняя реакция, называемая хлоргидринированием, имеет промышленное значение для получения хлоргидринов.
Структура образующихся продуктов в приведенных примерах сопряженного присоединения обусловлено несимметричным строением иона хлорония, благодаря которому положительный заряд в большей степени локализован на центральном атоме углерода и поэтому последний является объектом нуклеофильной атаки.
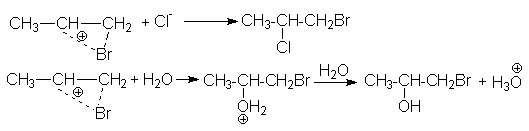
б) Взаимодействие с Н-электрофилами.
В качестве Н-электрофилов в этих реакциях могут выступать протонные кислоты, вода, сероводород, спирты, тиоспирты и др.
Присоединения Н-электрофилов к алкенам характеризуется стехиометрическим уравнением
R-CH=CH2 + HX R-CHX-CH3
В общем виде механизм этих реакций может быть проиллюстрирован последовательностью стадий:
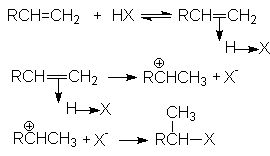
Скорость определяющей стадией этих реакций является образование -комплеса (карбкатиона). Это согласуется с кинетическим уравнением присоединения Н-электрофилов к алкенам
r=k[>C=C<][HX]
Характер скорость-определяющей стадии дает обоснование реакционной способности реагентов в этой реакции
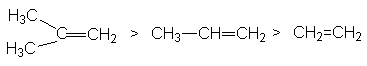
определяемой рядом стабильности карбкатионов
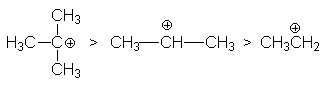
С другой стороны, ряд реакционной способности галогеноводородов
HI > HBr > HCl > HF
определяется кислотностью этих реагентов. Действительно, из рассмотрения скорость-определяющей стадии можно видеть, что при образовании -комплексов реализуется кислотность этих реагентов.
В случае Н-электрофилов, обладающих слабой кислотностью (Н2О, ROH, RSH и др.) для эффективного осуществления реакций присоединения используется кислотный катализ и более жесткие условия реакций. Так, например, реакции гидратации алкенов, осуществляемые в промышленном масштабе (синтез этанола и изопропанола), проводит в паровой фазе при температура около 300оС, давлениях 5-7мПа в присутствии фосфорной кислоты на Al2O3 или другом носителе. Роль катализатора может выполнять -Al2O3, содержащий протонные кислотные центры. Механизм этих реакций может быть проиллюстрирован на примере гидратации пропена
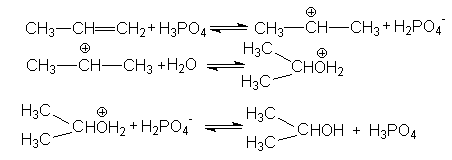
Суммирование левых и правых частей этих стадий приводит к стехиометрическому уравнению реакции гидратации
CH3CH=CH2 + H2O CH3CH(OH)CH3
Основная реакция осложняется протеканием двух побочных реакций:
полимеризации
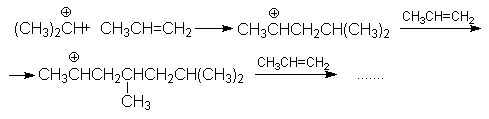
образование простых эфиров
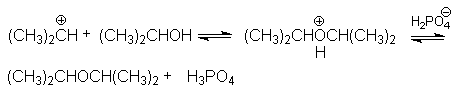
В кратком виде основную и побочные реакции можно представить системой конкурирующих реакций.
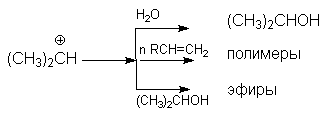
Из приведенной схемы видно, что факторами подавления побочных реакций являются: использование избытка водяного пара и снижение конверсии (концентрации спирта).
Порядок присоединения Н-электрофилов определяется правилом Марковникова, согласно которому водород Н-электрофила присоединяется к наиболее гидрированному атому. Причиной такого характера присоединения является большая энергетическая стабильность карбокатиона, образующегося при присоединении протона к более гидрированному углеродному атому двойной связи. Можно видеть, что дилемма образования карбокатионов в рассмотренном примере
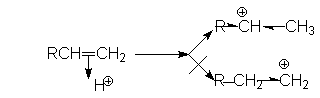
решается в пользу первого, стабилизированного индуктивным эффектом алкильных групп и эффектом снятия стерических напряжений. Соответственно, более предпочтительное образование карбокатиона R-CH -CH3 обусловливает образование продукта R-CHX-CH3.
В связи с этим современная трактовка правила Марковникова формулируется следующим образом: присоединение протона электрофила к алкену происходит в направлении образования наиболее стабильного карбкатиона. Такая формулировка объясняет, почему алкены, содержащие сильные электроноакцепторные заместители присоединяют Н-электрофилы против правила Марковникова в его классическом понимании.
CH2=CH-COOH + HBr |
|
Br-CH2CH2-COOH |
акриловая кислота |
|
-бромпропионовая кислота |
При взаимодействии акриловой кислоты с HBr можно представить два варианта присоединения протона.
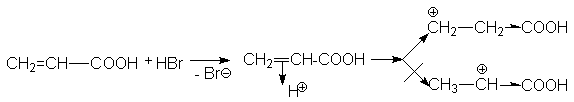
Можно видеть, что
второй карбкатион существенно
дестабилизирован электроноакцепторной
карбоксильной группой, поэтому его
образование энергетически невыгодно.
Предпочтительным является образование
карбкатиона ![]() ,
который и дает продукт
присоединения Br-CH2CH2COOH.
,
который и дает продукт
присоединения Br-CH2CH2COOH.
Обращение правила Мaрковникова имеет место и в случае свободнорадикального гидробромирования алкенов. Условием свободнорадикального процесса является использования инициаторов радикальных реакций, таких как пероксид водорода, органические гидропероксиды, пероксиды и др.
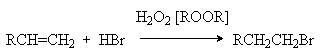
Характер образующегося продукта может быть обоснован в рамках свободнорадикального механизма гидробромирования.
H2O2 2HO
HO + HBr H2O + Br
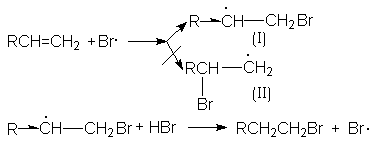
Можно видеть, что радикал I обладает большей стабильностью за счет действия двух эффектов: индуктивного и снятия стерических напряжений. Это обусловливает его предпочтительное образование и, что приводит к продукту присоединения против правила Мaрковникова.