

Термические методы анализа полимеров
Понятие термостойкости полимерных материалов довольно неоднозначно. С одной стороны, оно характеризует температурный интервал плавления или температуру размягчения полимера; с другой стороны, под термостойкостью понимают верхнюю предельную температуру, при которой в определенных условиях и при заданном времени выдержки не происходит существенных изменений механических или электрических свойств полимера. Тепло- и термостойкость полимеров связаны с их химическим строением и определяются физическими (температура плавления и температура стеклования) и химическими (стойкость к термической и термоокислительной деструкции) факторами. При кратковременном тепловом воздействии влияние оказывают факторы физические, в случае длительной термостойкости решающими являются химические факторы.
Для оценки термостойкости полимеров применяют экспресс-методы, позволяющие охарактеризовать температурные зависимости различных химических превращений и физических свойств, и длительные испытания. Данные, полученные с помощью экспресс-методов (термогравиметрического и дифференциально-термического анализа), дают ориентировочную оценку термостойкости. Наиболее часто используют термический анализ полимеров, который включает в себя термогравиметрию (ТГА), дифференциальный термический анализ (ДТА), дифференциальную сканирующую калориметрию (ДСК).
Термогравиметрический метод анализа
ТГА является широко распространенным стандартным методом анализа полимеров . Прибор для ТГА (дериватограф *) является термоаналитическим устройством, которое позволяет измерять изменение массы (ТГА) и скорость этого изменения (ДТГА) для одного образца, т.е. фиксировать интегральную и дифференциальную кривые потери его массы (рис. 1). Регистрируя во времени температуру и потерю массы образца, определяют температуру разложения и делают заключение о содержании веществ (например, наполнителя полимера).

Рис. 1. Графические результаты термогравиметрического анализа: 1 - кривая изменения массы образца (ТГ); 2 - изменение температуры
При нагревании образца в среде инертного газа изучают термическое разложение полимера, при нагревании на воздухе - его термоокислительную деструкцию. Используют навески полимеров около 0.1 г при нагреве до 1200 °С с различными скоростями, например 5 °С в минуту. В том случае если не происходит обратимых процессов выделения влаги или отщепления низкомолекулярных соединений в результате процессов циклизации, температура начала потери массы образца характеризует начало разложения материала. В качестве критерия термостабильности полимеров выбраны температуры 5, 10 и 50 %-ной потери массы на кривой ТГА .
Форма кривых ТГА зависит прежде всего от таких кинетических параметров, как порядок реакции, предэкспоненциальный множитель и энергия активации. Эти параметры имеют первостепенное значение для выяснения механизма термодеструкции полимера. Известно, что максимум дифференциальной кривой потери массы соответствует максимуму скорости любого процесса, протекающего при воздействии температуры, и равен 50 %-ной потере массы. Для определения энергии активации процесса термоокислительной деструкции используют метод Райха. При постоянной скорости нагревания RH значения энергии активации Е процессов разложения полимеров, а также порядок этой реакции п вычисляют по уравнению:
RT-d(Wi/W)/dt = (A/RH)-e-E/RT (Wi/W)n
или lgR7 = nlg (Wi/ W) - (E/2.303R) (1/T)
где RT - скорость реакции; WиWt- масса образца при температуре Т ; A - предэкспоненциальный множитель.
Величина (1/Т) постоянна, поэтому зависимость между lg Rr и lg (Wi/W) линейна; по ней определяют угол наклона прямой, равный порядку реакции п, и отрезок, отсекаемый прямой на оси ординат, характеризующий величину Е (энергию активации процесса термоокислительной деструкции). Считается, что термоокислительная деструкция полимеров протекает по реакции первого порядка.
В дифференциальном термотравиметрическом анализе посредством электронного дифференцирования импульсов непосредственно получают дифференциальную кривую, максимумы которой характеризуют температуры максимальной скорости деструкции.
* Дериватограф, представляет собой многофункциональную систему для термического анализа, позволяющую на одной ленте получить термогравиметрическую (ТГ—изменение массы образца при его нагревании), дифференциально-термическую (ДТА) и температурную (Т) кривые. С помощью дополнительных приспособлений можно также построить ТР- кривые (термического расширения), кривые производной ТР и ТГ, а также кривые выделенного газа.
Прибор, показанный на рис.2, включает аналитические весы, печь, устройство для регулирования температуры печи по заданной программе, тигли для образца и эталона, регулятор напряжения и гальванометрический самописец, работающий по принципу «световой луч — фотобумага». Аналитические весы с воздушным демпфированием балансира имеют точность 20±0.2 мг при предельном отклонении, рабочий интервал измерения массы — от 10 мг до 10 г. ТГП—кривая (производная ТГ) получается с помощью простого приспособления, состоящего из магнита и индукционной катушки. Магнит подвешивается на одном плече балансира. У обоих полюсов магнита размещены индукционные катушки. При измерении массы движение магнита индуцирует в катушках ток с напряжением, пропорциональным скорости изменения массы. Напряжение измеряется одним из гальванометров со световым лучом и записывается на ленту вместе с другими кривыми. Максимальная температура в печи 10000С. Печь может работать в атмосфере N2, CО2, O2 и других газов, но только при атмосферном давлении.
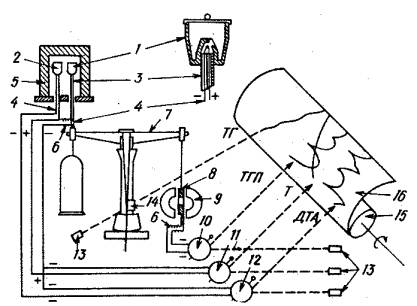
Рис.2. 1 – тигель для образца; 2 – тигель для инертного вещества; 3 – фарфоровая трубка; 4 – термопары; 5 – электрическая печь; 6 – нескручивающийся провод; 7 – весы; 8 – катушка; 9 – магнит; 10 – гальванометр для ТГП; 11 – гальванометр для измерения температуры; 12 – гальванометр для ДТА; 13 – лампы; 14 – оптическая щель; 15 – цилиндр для фоторегистрации; 16 – фотобумага.
Некоторые приборы отличаются тем, что в них скорость нагревания пробы непрерывно изменяется в зависимости от скорости изменения массы пробы. Это повышает разрешающую способность прибора и сокращает продолжительность анализа.
Метод ТГА имеет перед изотермическим методом следующие преимущества:
• требуется значительно меньше данных; температурная зависимость скорости потери массы образца может быть определена для различных температурных интервалов из результатов одного опыта, в то время как в изотермических методах для исследования каждой температурной области необходим отдельный образец;
поскольку для записи кривой ТГА требуется всего один образец исключаются возможные источники неточностей при изучении кинетики деструкции;
непрерывная запись потери массы и температуры позволяет учитывать специфику кинетики деструкции.
Недостатком метода ТГА является то обстоятельство, что потеря массы, обусловленная отщеплением газообразных продуктов деструкции, в отдельных случаях может компенсироваться увеличением массы при протекании процессов окислительной деструкции.
ТГА при высокой температуре не регистрирует реакций гидролиза, сопровождающихся образованием большого количества фрагментов разрушенных макромолекул. Поэтому необходимо проведение подробного анализа продуктов с использованием одного или нескольких аналитических методов. В комплексном методе нет необходимости в приготовлении пробы образца; во время нагревания образу через дериватограф проходит поток газа-носителя, уносящий продукты разложения полимера в камеру ИК-спектрометра.
Одна из возможных областей применения метода ТГА - определение структурных характеристик пористых материалов. Для этого измеряют количество жидкости, десорбирующейся из пористого материала, предварительно насыщенного этой жидкостью. Совместное рассмотрение интегральной и дифференциальной кривых потери массы образца при термодесорбции жидкости позволяет определять количество жидкости в порах, а также количество жидкости, адсорбированной на поверхности пор в виде монослоя. Удельный объем пор рассчитывают по формуле:
![]()
где тж - масса жидкости в порах материала, г (находится из дериватограммы); тобр - масса образца, г; рж - плотность жидкости, г/см3.
Удельную поверхность пор (м2/г) находят по формуле:
![]()
где а - масса жидкости в монослое, г; та - масса молекул жидкости, г, Sa - площадь молекулы жидкости, м2 (площадь молекулы бензола 0.43-10-18 м2, воды - 0,22-10 -18 м2). При относительной ошибке определения, сравнимой с точностью других известных методов, продолжительность испытаний намного меньше и составляет 40-50 минут. Очевидно, данный метол может оказаться полезным при оценке свойств наполнителей, применяемых при переработке полимеров.