

4.4. Закон Гесса
Пользуясь
табличными значениями
![]() и
,
можно рассчитать энтальпии различных
химических процессов и фазовых
превращений. Основанием для таких
расчетов является закон
Гесса,
сформулированный петербургским
профессором Г.
И. Гессом
(1841 г.): «Тепловой
эффект (энтальпия) процесса зависит
только от начального и конечного
состояния и не зависит от пути перехода
его из одного состояния в другое».
и
,
можно рассчитать энтальпии различных
химических процессов и фазовых
превращений. Основанием для таких
расчетов является закон
Гесса,
сформулированный петербургским
профессором Г.
И. Гессом
(1841 г.): «Тепловой
эффект (энтальпия) процесса зависит
только от начального и конечного
состояния и не зависит от пути перехода
его из одного состояния в другое».
Анализ закона Гесса позволяет сформулировать следующие следствия:
Энтальпия реакции равна разности сумм энтальпий образования конечных и начальных участников реакций с учетом их стехиометрических коэффициентов.
ΔH = ΣΔHобр.конечн – ΣΔHобр.нач
Энтальпия реакции равна разности сумм энтальпий сгорания начальных и конечных реагентов с учетом их стехиометрических коэффициентов.
ΔH = ΣΔHсгор.нач – ΣΔHсгор.конечн
Энтальпия реакции равна разности сумм энергий связей Eсв исходных и конечных реагентов с учетом их стехиометрических коэффициентов.
В ходе химической реакции энергия затрачивается на разрушение связей в исходных веществах (ΣEисх) и выделяется при образованиии продуктов реакции (–ΣEпрод). Отсюда
ΔH° = ΣEисх – ΣEпрод |
Следовательно, экзотермический эффект реакции свидетельствует о том, что образуются соединения с более прочными связями, чем исходные. В случае эндотермической реакции, наоборот, прочнее исходные вещества.
При определении энтальпии реакции по энергиям связей уравнение реакции пишут с помощью структурных формул для удобства определения числа и характера связей.
Энтальпия реакции образования вещества равна энтальпии реакции разложения его до исходных веществ с обратным знаком.
ΔHобр = –ΔHразл |
Энтальпия гидратации
равна разности энтальпий растворения
безводной соли
![]() и
кристаллогидрата
и
кристаллогидрата
![]()
Из вышесказанного видно, что закон Гесса позволяет обращаться с термохимическими уравнениями как с алгебраическими, т. е. складывать и вычитать их, если термодинамические функции относятся к одинаковым условиям.
Например, диоксид углерода можно получить прямым синтезом из простых веществ (I) или в две стадии через промежуточный продукт (II):
|
Рисунок 4.1. Энтальпия первого пути равна сумме энтальпий отдельных стадий второго пути. |
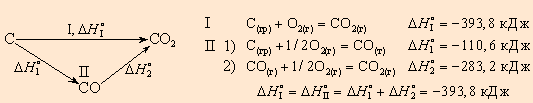
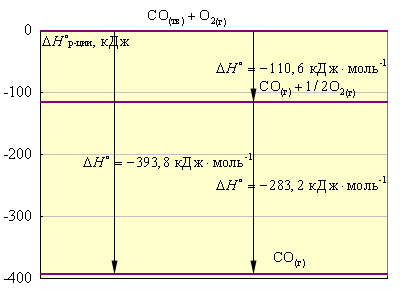
Эти термохимические реакции можно представить в виде энтальпийных диаграмм. Естественно, за начало следует принять стандартные состояния простых веществ, энтальпии которых равны нулю. Образование сложных веществ (CO и CO2) сопровождается понижением энтальпии системы.
|
Рисунок 4.2. Энтальпийная диаграмма C + O2. |
4.5. Энтропия
Изменение энтальпии системы не может служить единственным критерием самопроизвольного осуществления химической реакции, поскольку многие эндотермические процессы протекают самопроизвольно. Иллюстрацией этого служит растворение некоторых солей (например, NH4NO3) в воде, сопровождающееся заметным охлаждением раствора. Необходимо учитывать еще один фактор, определяющий способность самопроизвольно переходить из более упорядоченного к менее упорядоченному (более хаотичному) состоянию.
Энтропия (S) – термодинамическая функция состояния, которая служит мерой беспорядка (неупорядоченности) системы. Возможность протекания эндотермических процессов обусловлена изменением энтропии, ибо в изолированных системах энтропия самопроизвольно протекающего процесса увеличивается ΔS > 0 (второй закон термодинамики).
Л. Больцман определил энтропию как термодинамическую вероятность состояния (беспорядок) системы W. Поскольку число частиц в системе велико (число Авогадро NA = 6,02∙1023), то энтропия пропорциональна натуральному логарифму термодинамической вероятности состояния системы W:
|
|
|
Размерность энтропии 1 моля вещества совпадает с размерностью газовой постоянной R и равна Дж∙моль–1∙K–1. Изменение энтропии *) в необратимых и обратимых процессах передается соотношениями ΔS > Q / T и ΔS = Q / T. Например, изменение энтропии плавления равно теплоте (энтальпии) плавления ΔSпл = ΔHпл/Tпл Для химической реакции изменение энтропии аналогично изменению энтальпии
|
|
|
*) термин энтропия был введен Клаузиусом (1865 г.) через отношение Q/T (приведенное тепло).
Здесь ΔS° соответствует энтропии стандартного состояния. Стандартные энтропии простых веществ не равны нулю. В отличие от других термодинамических функций энтропия идеально кристаллического тела при абсолютном нуле равна нулю (постулат Планка), поскольку W = 1.
Энтропия вещества или системы тел при определенной температуре является абсолютной величиной. В табл. 4.1 приведены стандартные энтропии S° некоторых веществ.
Соединение
|
||||||||||||||||||||||||||||||||||||||||||||
Таблица 4.1. Стандартные энтропии некоторых веществ. |
Из табл. 4.1 следует, что энтропия зависит от:
Агрегатного состояния вещества. Энтропия увеличивается при переходе от твердого к жидкому и особенно к газообразному состоянию (вода, лед, пар).
Изотопного состава (H2O и D2O).
Молекулярной массы однотипных соединений (CH4, C2H6, н-C4H10).
Строения молекулы (н-C4H10, изо-C4H10).
Кристаллической структуры (аллотропии) – алмаз, графит.
Наконец, рис. 4.3 иллюстрирует зависимость энтропии от температуры.
|
Рисунок 4.3. Зависимость энтропии от температуры для свинца: ΔSпл = 8 Дж·моль–1·К–1; Tпл = 600,5 К; ΔSкип = 88 Дж·моль–1·К–1; Tкип = 2013 К. |
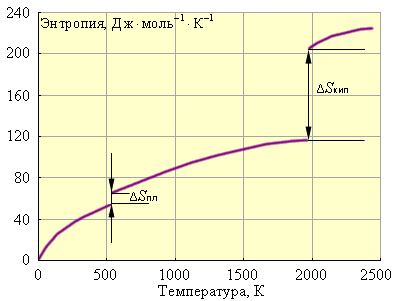
Следовательно, стремление системы к беспорядку проявляется тем больше, чем выше температура. Произведение изменения энтропии системы на температуру TΔS количественно оценивает эту тендецию и называется энтропийным фактором.