

Инфекционные болезни
Инфекционные болезни - одна из главных проблем здравоохранения во всем мире; они характеризуются высоким уровнем заболеваемости и смертности. С генетической точки зрения важно, что они отличаются от неинфекционных многофакторныхных болезней тем, что имеют специфическую причину - возбудителя болезни (патоген). Однако развитие болезни, характер течения инфекционного процесса, чувствительность организма к возбудителю определяются сложным взаимодействием факторов окружающей среды, патогена и наследственных факторов «хозяина» (рис. 6.5).
Основной взгляд на генетику подверженности распространенным инфекциям (малярии, ВИЧ/СПИДу, микобактериальным и вирусным инфекциям) обозначается как «одна инфекция - множество генов». Однако у лиц с первичными иммунодефицитами часто развиваются самые разнообразные инфекционные заболевания, которые рассматриваются как осложненное течение моногенного иммунодефицита. Такие случаи обозначаются как «один ген - многочисленные инфекции».
В настоящее время активно осуществляется поиск локусов (генных вариантов) чувствительности к инфекционным заболеваниям. Основной подход в этих исследованиях - скрининг генов-кандидатов и анализ их ассоциаций с инфекционными болезнями. К решению этой задачи в последнее время привлекаются и полногеномные ассоциативные исследования.
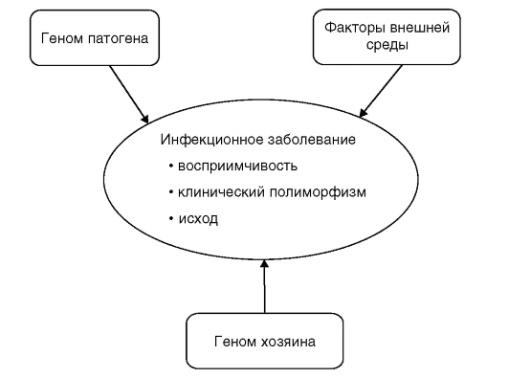
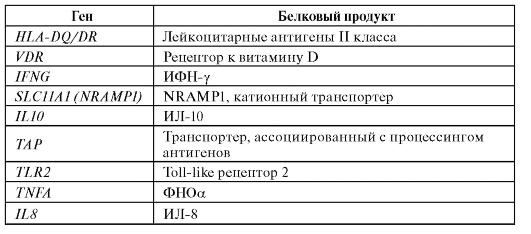
Рис. 6.5. Триединство факторов патогенетики инфекционного процесса
В отношении туберкулеза, наиболее активно исследуемого с генетических позиций заболевания, накоплена достаточно большая информация (табл. 6.10.).
Таблица 6.10. Гены, для которых показана ассоциация с туберкулезом
Окончание таблицы 6.10

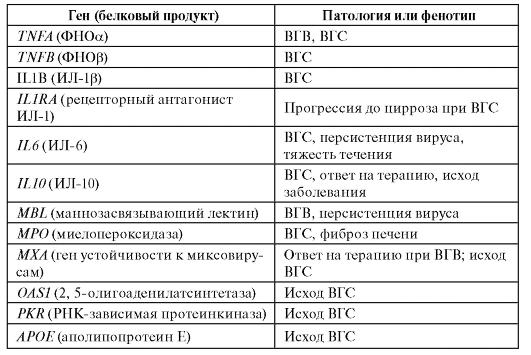
В таблице 6.11 приведены результаты ассоциативных исследований для вирусных гепатитов В и С.
Таблица 6.11. Гены, для которых показана связь с вирусным гепатитом В и С и ассоциированными клиническими фенотипами
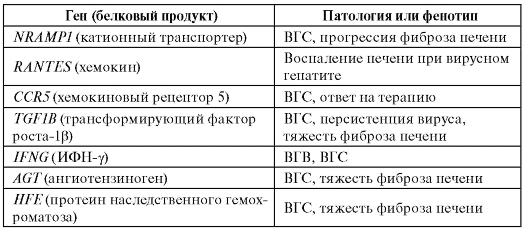
Примечание. ВГВ - вирусный гепатит B; ВГС - вирусный гепатит C.
Генетика инфекционных заболеваний, еще совсем недавно считавшаяся мало перспективной в силу небольших коэффициентов наследования (h2 - 10-20%) этой патологии, сегодня активно исследуется всем современным арсеналом генетических методов.
Злокачественные новообразования
Среди многочисленных и широко распространенных многофакторныхных болезней особую группу составляют злокачественные новообразования.
С позиций генетической классификации заболеваний человека большинство злокачественных новообразований относится к болезням, обусловленным мутациями в соматических клетках. Наряду с этим существуют и наследственные формы опухолей, обязательным компонентом развития которых является наследование генной мутации через половые клетки родителей (иногда их называют герминативными). Подробно о наследственной предрасположенности к онкологическим заболеваниям см. в статье Е.Н. Имянитова на компакт-диске.
Следует подчеркнуть, что злокачественные опухоли - болезни соматических клеток, способных к делению (стволовые клетки, клетки-предшественники). Опухоль возникает в результате накопления различных типов генетических нарушений, приводящих к потере геномного контроля над процессами деления, роста и естественной убыли клеток. Предполагается, что для индукции солидных типов рака необходимо накопление не менее 5-7 мутаций. Развитие
злокачественных опухолей протекает через 3 основные стадии: инициацию, промоцию и прогрессию, при этом скорость данных процессов определяется частотой возникновения мутаций, численностью клеточной популяции, скоростью пролиферации и преимуществом в размножении мутантных клеток.
Выделяют 3 основные группы факторов риска возникновения злокачественных новообразований. Первую из них составляют физические факторы, а именно ионизирующее и ультрафиолетовое излучение, повышающее частоту генных мутаций в соматических клетках.
Вторую группу составляют химические факторы, к которым относятся канцерогены - широкий спектр химических соединений органической и неорганической природы, способных вызывать мутации в соматических клетках (мутагены, или опухолевые инициаторы) или индуцировать опухолевый рост клеток, подвергшихся действию мутагена (митогены, или опухолевые промоторы). Рак возникает в результате последовательного действия инициатора и промотора, при этом риск злокачественной трансформации клеток зависит от дозы канцерогенов, продолжительности и периодичности их воздействия.
Следующей группой факторов риска являются биологические. Среди них особое место занимают вирусы. ДНК-содержащие вирусы, представители семейств паповавирусов (папилломавирус), гепаднавирусов (вирус гепатита B) и герпес-вирусов (вирус Эпстайна-Барр) способны к интеграции в геном клетки-хозяина и к индукции незапланированных циклов ее репликации. Многие вирусные онкогены способны при этом блокировать ключевые опухолесупрессорные механизмы клетки. РНК-содержащие вирусы семейства ретровирусов (вирус Т-лейкоза человека I типа и ВИЧ) имеют геном, представленный двумя молекулами одноцепочечной РНК. В геноме этих вирусов закодирован фермент обратная транскриптаза, необходимый для синтеза копий вирусной ДНК. Встраивание вирусной ДНК в геном клетки-хозяина - обязательный этап жизненного цикла ретровируса. Подобное встраивание может сопровождаться инсерционным мутагенезом, ведущим к появлению структурных мутаций в кодирующих последовательностях генов.
|
|
Среди биологических факторов риска отдельное место, несомненно, занимает наследственная предрасположенность к злокаче-
ственным новообразованиям, молекулярные основы которой будут рассмотрены ниже.
На каждом из этапов формирования опухоли развертываются скоординированные в патологическом направлении процессы сначала на молекулярном, а затем на клеточном уровне. Рассматривая канцерогенез как результат нарушения геномного контроля баланса численности клеточной популяции, можно выделить две группы генов, мутации которых этиологически связаны с развитием рака. Речь идет о протоонкогенах и опухолесупрессорах.
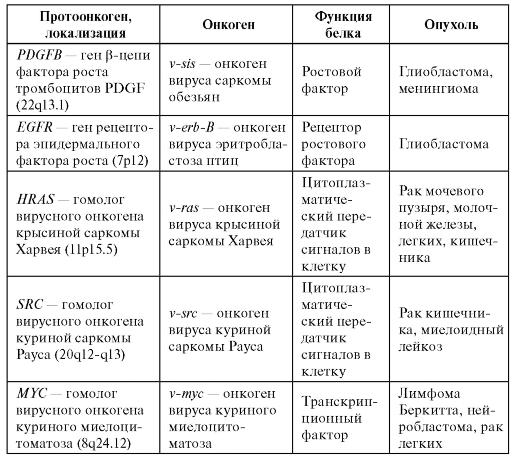
Протоонкогены (первая группа) - нормальные клеточные гены, контролирующие рост и размножение клеток. Продукты протоонкогенов осуществляют контроль роста и деления клеток и представлены ростовыми факторами и их рецепторами, внутриклеточными передатчиками сигналов, факторами транскрипции. Измененные в результате мутации протоонкогены или гены, вносимые в геном некоторыми вирусами, получили название онкогенов. Онкогены оказывают стимулирующие влияние на клеточную пролиферацию. Их действие носит доминантный характер, т.е. для развития рака достаточно мутации в одном из аллелей онкогена.
Было замечено, что некоторые вирусы приводят к развитию злокачественных опухолей у животных. Поскольку вирус привносит в клетку хозяина свой генетический материал, то предположили, что геномы вирусов содержат онкогены. В конце 70-х годов XX в. в РНК-содержащем вирусе куриной саркомы Рауса был выделен участок РНК, ответственный за злокачественную трансформацию клеток. Так был открыт первый онкоген - v-src. Позднее оказалось, что в нормальных клетках есть гены, по своей структуре близкие к онкогенам - протоонкогены. По всей видимости, в ходе эволюции вирусные онкогены возникли из нормальных клеточных генов в результате рекомбинации между предковым нетрансформирующим вирусом и ДНК клетки-хозяина. В нормальных клетках протоонкогены находятся под контролем других клеточных генов. Интеграция вируса в геном клетки-хозяина освобождает их от этого контроля, приводит к нерегулируемой активации и превращению в онкоген. К настоящему времени идентифицировано более 100 протоонкогенов и их число продолжает расти. Характеристика некоторых протоонкогенов и онкогенов приведена в табл. 6.12.
Таблица 6.12. Характеристика протоонкогенов и онкогенов
В клетке протоонкогены могут трансформироваться в онкогены через следующие механизмы.
- Рекомбинация с ретровирусной ДНК. Интеграция вирусной нуклеиновой кислоты в геном клетки-хозяина имеет два последствия. Во-первых, это нарушение структуры гена, в который произошло встраивание чужеродного генетического материала (инсерционный или вставочный мутагенез). Во-вторых, это стимулирование незапланированных циклов репликации ДНК и клеточного деления.
- Хромосомные транслокации. Перемещение участка хромосомы, содержащего протоонкоген, на другую хромосому может привести либо к изменению структуры гена, либо к нарушению
регуляции его активности. Например, транслокация t(9;22) (q34;q11), или так называемая «филадельфийская хромосома», встречается у 95% больных хроническим миелоидным лейкозом (рис. 6.6). Она возникает в клетках костного мозга еще до проявления основных симптомов заболевания и имеет важное прогностическое значение. При этой перестройке участок длинного плеча хромосомы 22 транслоцирован на длинное плечо хромосомы 9, а небольшой фрагмент хромосомы 9, содержащий ген ABL, присоединен к участку хромосомы 22, содержащему ген BCR. В результате транслокации на хромосоме 22 образуется химерный ген ABL-BCR, продукт которого (тирозиновая протеинкиназа) стимулирует непрерывное клеточное деление. Важно отметить, что расшифровка цитогенетических и молекулярных механизмов формирования химерного белка позволила создать новый лекарственный препарат (иматиниб), являющийся ингибитором тирозиновых протеинкиназ и демонстрирующий обнадеживающие результаты в лечении больных хроническим миелолейкозом. Другим примером, демонстрирующим роль хромосомных перестроек в активации протоонкогенов, является реципрокная транслокация t(8;14)(q24;q32), выявляемая при лимфоме Беркитта. Транслокация отделяет протоонкоген C-MYC (8q24.12) от его
нормального промотора. В новом месте он попадает под сильный регуляторный элемент гена иммуноглобулина H, расположенного на хромосоме 14. В результате перестройки в клетке резко повышается продукция белка C-MYC - транскрипционного фактора, обладающего онкогенными свойствами. - Амплификации протоонкогенов. Этот процесс существенно увеличивает число копий протоонкогена в клетке, что приводит к
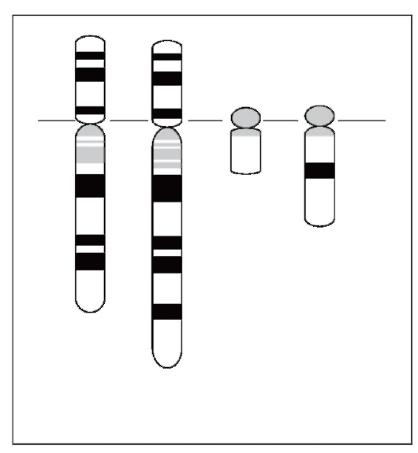
Рис. 6.6. Транслокация t(9;22) при хроническом миелолейкозе
образованию большого количества соответствующих онкопротеинов. Часто амплификация специфических протоонкогенов является признаком определенных опухолей (например, при мелкоклеточном раке легкого обнаруживается амплификация C-MYC, N-MYC и L-MYC протоонкогенов). - Точковые мутации в кодирующих последовательностях протоонкогенов приводят к синтезу онкогенных белков. Наряду с генными мутациями может наблюдаться его существенная амплификация в онкогене, что усиливает трансформирующие клетку свойства.
Вторая группа генов, мутации в которых приводят к развитию злокачественных новообразований, - опухолесупрессорные гены. Их функция заключается в ингибировании клеточного деления. В отличие от протоонкогенов, действие опухолесупрессоров рецессивно, т.е. для развития опухоли необходимо наличие мутации в обоих аллелях одного гена. С мутациями опухолесупрессорных генов связаны наследственные формы новообразований, обусловленные комбинацией герминативных и соматических мутаций. Некоторые опухолесупрессоры обладают свойством так называемых мутаторных генов - их инактивация обусловливает резкое возрастание частоты мутаций в других локусах генома. Как правило, продукты таких генов вовлечены в регуляцию процессов репликации и репарации ДНК, клеточного цикла и апоптоза.
|
|
Механизмы действия опухолесупрессорных генов были открыты при исследовании ретинобластомы - злокачественной опухоли сетчатки глаза у детей. Впервые ретинобластома как самостоятельное заболевание с аутосомно-доминантным типом наследования была описана еще в 1902 г. В 1971 г. А. Кнудсен предположил, что для возникновения болезни необходимы мутации в каждой копии гена, отвечающего за развитие опухоли.
Семейные случаи ретинобластомы (примерно 40% всех случаев заболевания) характеризуются более ранним возрастом начала болезни, предрасположенностью к другим типам злокачественных опухолей и являются в основном двусторонними или мультифокальными. Согласно предположению А. Кнудсена, семейные формы возникают в результате наследования одной мутации от родителей, а вторая мутация возникает уже в соматических клетках в нормальном аллеле. Шанс второй мутации достаточно высок, что и приводит к «доминантной предрасположенности» к развитию опухоли.
Спорадические случаи ретинобластомы возникают в результате двух соматических мутаций. Для них характерен поздний возраст начала заболевания, а также отсутствие предрасположенности к другим типам злокачественных новообразований. Как правило, спорадическая ретинобластома односторонняя (имеет один фокус возникновения).
Данные рассуждения А. Кнудсена легли в основу его «двухударной гипотезы» канцерогенеза, нашедшей хорошее подтверждение для большинства наследственных форм злокачественных новообразований (рис. 6.7). Индивид наследует от одного из родителей мутацию в опухолесупрессорном гене. Это может быть точковая мутация или микроделеция. Гетерозиготность индивида по данному локусу страхует его от возникновения опухоли. Однако в течение жизни в соматических клетках может возникнуть мутация и в нормальном аллеле опухолесупрессорного гена. Это приведет к потере гетерозиготности и, как результат, к полному отсутствию продукта опухолесупрессорного гена в клетке. Очевидно, что подобные события (две мутации) могут возникать и у индивидов, не несущих герминативных мутаций, однако вероятность их возникновения будет значительно ниже, поскольку
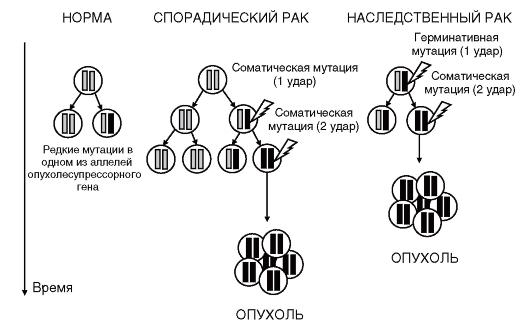
Рис. 6.7. «Двухударная гипотеза» канцерогенеза Кнудсена
требуется последовательное повреждение обоих нормальных аллелей конкретного опухолесупрессорного гена.
Ген ретинобластомы (RB1) стал первым открытым опухолесупрессорным геном у человека. Это открытие было сделано в 1986 г., т.е. спустя 15 лет после формулирования «двухударной гипотезы». Ген RB1 локализован в сегменте 13q14 и кодирует ретинобластомный белок (pRB) - негативный регулятор клеточного цикла. В гипофосфорилированном состоянии pRB связывает семейство транскрипционных факторов E2F, подавляя таким образом деление клетки. В нормальных условиях запуск клеточного деления осуществляется комплексом циклинов и циклинзависимых киназ, которые фосфорилируют pRB и освобождают тем самым факторы транскрипции. Точковые мутации в гене RB1 или аберрантное гиперметилирование его промоторного региона, а также микроделеции участка хромосомы 13q14 приводят к отсутствию опухолесупрессорного белка. В результате клетки, имеющие повреждения и в других локусах генома, не останавливаются в своем размножении, что увеличивает шансы развития опухолевого процесса.
У человека описан ряд злокачественных новообразований, возникающих за счет потери гетерозиготности (табл. 6.13). Для многих форм известны не только хромосомная локализация опухолесупрессорного гена, но и его структура, мутации и первичные продукты. Часто для возникновения одной и той же опухоли необходима потеря гетерозиготности не в одном, а в нескольких локусах. Кроме того, нужны еще мутации в онкогенах. Многокомпонентность генетических событий канцерогенеза очевидна.
Таблица 6.13. Опухоли, возникающие в связи с потерей гетерозиготности
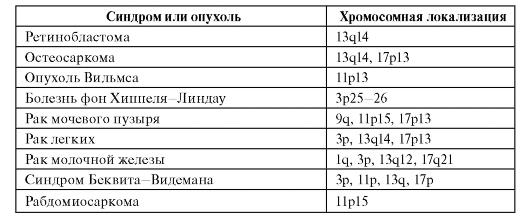
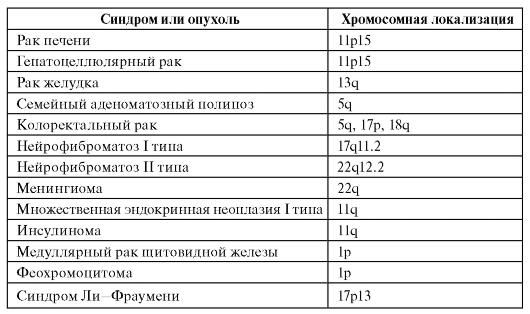
Окончание таблицы 6.13
Злокачественные новообразования, развивающиеся на основе унаследованных мутаций, иногда имеют характер семейных синдромов. Признаки таких синдромов:
|
|
• высокая частота опухолей у родственников I и II степени родства;
• наличие в родословной несколько близких родственников со сходными формами опухолей, например, молочной железы и яичника, кишечника, эндометрия;
• наличие двух членов семьи со сходными редкими формами опухолей;
• необычно ранний возраст начала заболевания;
• двусторонние опухоли парных органов;
• синхронность или непрерывность возникновения опухолей;
• опухоли органов разных систем у одного индивида.
Одним из примеров таких семейных форм злокачественных новообразований является синдром Ли-Фраумени, впервые описанный в 1988 г. как заболевание с аутосомно-доминантным типом наследования. Диагноз основывается на нахождении от 2 до 6 типов опухолей в одной родословной и более. Саркомы начинаются в возрасте до 5 лет, остеосаркомы - в юношеском возрасте, а опухоль мозга, молочной железы, аденокарцинома желудка или лейкемия проявляются до 30-летнего возраста. В 1990 г. было показано, что больные
имеют мутации в гене-супрессоре опухоли TP53 (17p13.1). В норме продукт данного гена белок p53 практически не обнаруживается в клетках вследствие чрезвычайно короткого времени полураспада. Функция p53 заключается в контроле целостности ДНК, регуляции клеточного цикла и апоптоза. Белок определяет, произошла ли репарация повреждений ДНК в клетке. Если повреждение не может быть репарировано, то запускается программа апоптоза. При повреждении ДНК p53 активируется и стимулирует транскрипцию гена p21, который кодирует ингибитор циклинзависимой киназы. Белок p21 присоединяется к комплексу циклина и циклинзависимой киназы и инактивирует его. В результате клетка останавливается в G1-фазе клеточного цикла. Очевидно, что мутации в гене TP53 делают невозможным прохождение такого каскада реакций и увеличивают шанс злокачественной трансформации клеток. Мутации данного гена найдены более чем в 50% разных форм злокачественных новообразований. Скрининг мутаций гена TP53 имеет важное клиническое значение для прогноза как в семьях с синдромом Ли-Фраумени, так и при спорадических формах опухолей.
Около 5-10% случаев рака молочной железы составляют наследственные формы, обусловленные передачей потомству мутантных вариантов высокопенетрантных генов репарации ДНК и апоптоза. Особое место среди них занимают гены BRCA1 (17q21) и BRCA2 (13q12). Продукты этих генов активируются в ответ на повреждение ДНК и вместе с белком RAD51, вовлеченным в осуществление гомологичной рекомбинации, формируют комплекс, участвующий в репарации двухцепочечных разрывов ДНК. Открытие генов BRCA1 и BRCA2 имеет большое значение для прогноза возникновения наследственных форм рака молочной железы. Женщины, несущие мутации этих генов, имеют высокий риск развития опухолей молочной железы и яичников. Этот риск особенно повышается в семьях, где есть два случая рака молочной железы и более, а также отмечается раннее начало заболевания - в возрасте до 40 лет.
Наиболее распространенной из наследственных форм опухолей кишечника является наследственный неполипозный колоректальный рак. В его развитии существенную роль играют мутации в генах мисс-матч репарации ДНК - MSH2 (2p11), MSH6 (2p22), MLH1 (3p21.3), MLH3 (14q24.3), PMS1 (2q31) и PMS2 (7p22). Инактивация обоих аллелей одного из этих генов приводит к резкому накоплению ошибок репликации ДНК (гены с мутаторным эффектом), что выявляется
по высокой изменчивости числа копий микросателлитных повторов. Подобный феномен микросателлитной нестабильности, помимо наследственного колоректального рака, наблюдается примерно в 10-20% случаев спорадических опухолей разных типов, свидетельствуя о том, что дефекты в генах репарации ДНК - один из общих механизмов канцерогенеза.
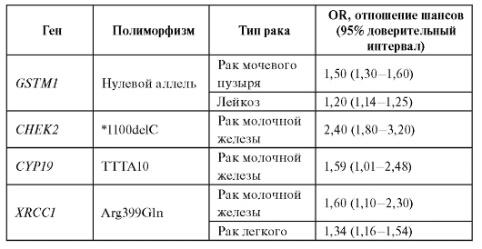
Рассмотренные выше события канцерогенеза не исчерпывают всей многокомпонентности генетической предрасположенности к раку и многофакторности причин развития опухолевого процесса. Можно определить еще целый ряд наследственных характеристик индивида, имеющих отношение к канцерогенезу. Так, например, метаболизм канцерогенов в клетках определяется биохимическими системами, многие из которых генетически полиморфны. Полиморфизм активирующих ксенобиотики ферментов (эстеразы, оксигеназы, изоформы цитохрома Р450) и детоксицирующих ферментов (различные трансферазы) определяе т индивидуальную чувствительность организма к канцерогенным воздействиям (см. главу 7).
Метаанализ данных литературы показывает, что наследственный полиморфизм генов, вовлеченных в биотрансформацию ксенобиотиков, демонстрирует наиболее значимые ассоциации с риском развития онкологических заболеваний. Некоторые примеры ассоциаций приведены в табл. 6.14.
Таблица 6.14. Ассоциации генетических полиморфизмов с различными типами опухолей
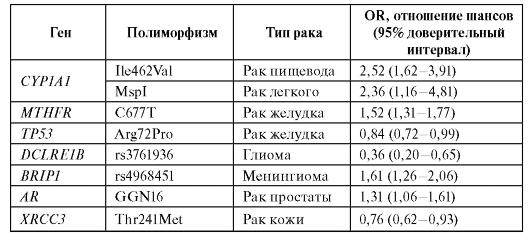
Окончание таблицы 6.14
Таким образом, медико-генетическое консультирование семьи с наследственной предрасположенностью к злокачественным новообразованиям - сложнейшая задача. Требуются специальная подготовка врачей-генетиков в вопросах генетических основ канцерогенеза и хорошая лабораторная база. Только такое сочетание позволяет правильно выявить предрасположенность членов семьи, определить необходимость их диспансерного наблюдения и характер профилактических мероприятий. С молекулярно-биологическими технологиями в современной онкологии можно ознакомиться в одноименной статье С.П. Коваленко на компакт-диске.
ЗНАЧЕНИЕ НАСЛЕДСТВЕННОЙ
ПРЕДРАСПОЛОЖЕННОСТИ В ОБЩЕЙ ПАТОЛОГИИ
ЧЕЛОВЕКА И КЛИНИЧЕСКОЙ ПРАКТИКЕ
Идентификация генетических вариантов подверженности широко распространенным заболеваниям, обозначаемая иногда как генетическое тестирование многофакторного заболевания, является активно развивающейся областью исследований, которая имеет важное теоретическое и практическое значение. Число публикаций по генетическим ассоциациям ежегодно в последнее десятилетие удваивается, и эта информация излагается в 1500 научных журналах на различных языках. Направления, по кото-
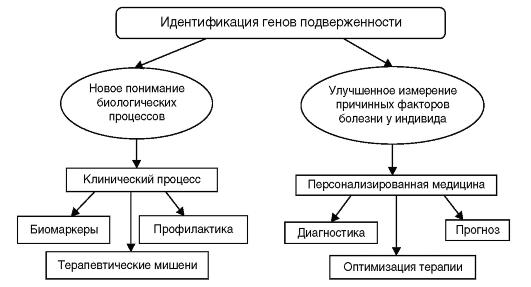
Рис. 6.8. Применение результатов исследования генетических ассоциаций
рым осуществляется систематизация накапливаемой информации, представлены на рис. 6.8.
Полногеномные ассоциативные исследования, анализирующие одновременно до 1 млн геномных вариантов, раскрывают биологические основы многофакторных заболеваний, открывая новые, до сих пор неизвестные метаболические пути формирования патологических фенотипов (болезней), обнаруживая терапевтические мишени, что способствует созданию новых лекарственных средств. Идентификация биомаркеров позволяет надеяться, что риск заболевания может быть снижен путем проведения оптимальных схем лечения. Даже умеренные ассоциации «генотип-фенотип» могут быть использованы для более широких возможностей перехода от теории к практике.
Однако следует заметить, что большинство идентифицированных к настоящему времени ассоциаций генетических полиморфизмов с многофакторными болезнями объясняют небольшой процент (2-10%) индивидуальной вариабельности в риске заболевания. В связи с этим, прежде чем «генетические профили» станут пригодны к широкому использованию в клинической практике, необходимы дополнительные уточняющие исследования и разработки. Они касаются совершенствования подходов к расчету рисков заболеваний, создания правового обоснования для применения генетических тестов, согла-
сованных действий исследователей, врачей и пациентов. Клиническая практика должна опираться на доказательную медицину. Результаты генетического тестирования многофакторного заболевания никогда не будут единственными ориентирами в принятии решений по лечебно-диагностическим и профилактическим мероприятиям - генетические тесты не вместо, а вместе с фенотипическими маркерами составляют основу в персонализированном прогнозе, всегда вероятностном.