

3.2.3. Аномалія в’язкості за сталого плину
Явище аномалії в’язкості полягає в тім, що ефективна в’язкість зі збільшенням τ і звичайно зменшується. Це явище має великий практичний інтерес, як основна характеристика гідродинамічної поведінки полімерних систем у реальних умовах підчас здійснення процесів переробки.
В умовах прояву
аномалії в’язкості режими сталої
зсувної плину описують функцією
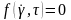 ,
графічне зображення якої називають
«кривою плину».
,
графічне зображення якої називають
«кривою плину».
|
Рис. 3.5. Повна крива плину полімерів |
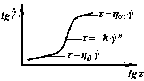
 й
,
що відповідає лінійним режимам
ньютонівського плину. Для розплавів
полімерів звичайно одержують тільки
обмежену ділянку повної кривої. У
вузькому інтервалі змінних криві плину
можуть бути апроксимовані прямою (у
подвійних логарифмічних координатах)
з кутовим коефіцієнтом «n»,
що має назву індекс
плину.
й
,
що відповідає лінійним режимам
ньютонівського плину. Для розплавів
полімерів звичайно одержують тільки
обмежену ділянку повної кривої. У
вузькому інтервалі змінних криві плину
можуть бути апроксимовані прямою (у
подвійних логарифмічних координатах)
з кутовим коефіцієнтом «n»,
що має назву індекс
плину.
Всі теорії аномалії в’язкості в полімерних системах можна умовно розділити на наступні великі групи:
- теорії, що зв’язують явище аномалії в’язкості із впливом напруження або швидкості зсуву на величину потенційного бар’єра, що перешкоджає переходу молекулярно-кінетичних одиниць із одного рівноважного стану в інший;
- структурні теорії аномалії в’язкості, що розглядають залежність ефективної в’язкості від режиму плину як результат оборотного (тиксотропного) руйнування-відновлення надмолекулярної структури полімеру;
- гідродинамічні теорії, у яких зменшення ефективної в’язкості полімерного матеріалу унаслідок підвищення швидкості зсуву пояснють зміною форми і, відповідно, опору переміщенню макромолекул.
Однак, всі ці теорії описують аномалію в’язкості переважно з якісного боку. Відсутність теорії аномалії в’язкості, що добре погоджується з кількісними експериментальними даними, породила безліч емпіричних формул, найбільш важливою з яких є статечне рівняння виду:
|
(3.3) |

де k − коефіцієнт консистенції; n − індекс плину.
В інтервалі величин напружень і швидкостей зсуву, що мають найбільше значення в технології переробки полімерних матеріалів, рівняння (3.3) досить добре описує поведінку багатьох полімерних систем. Крім того, воно зручне з огляду простоту графічного визначення індексу плину і придатність для розрахунків підчас рішень різних прикладних задач.
Величина початкової в’язкості має величезне значення для характеристики властивостей полімерних систем. Тому, у тих випадках, коли область практично постійної найбільшої в’язкості експериментально недосяжна, важливим стає метод її знаходження екстраполяцією залежності ln η = f(τ) до значення τ → 0. Помилка в цьому випадку не перевищує ± 10 %.
|
Рис. 3.6. Екстраполяція залежності lnη = f(τ) |

За деякого граничного значення M/Mкр ≈ 5 у полімері проявляється здатність переходити у високоеластичний стан, для якого характерно існування тривимірної сітки взаємозалежних макромолекул. Це є суто релаксаційний перехід, що здійснюється в ізотермічних умовах при Т > Тс. Він проявляється найбільш чітко в полімерах з вузьким молекулярно-масовим розподілом. У разі збільшення швидкості зсуву в ізотермічних умовах може спостерігатися перехід полімеру з текучого у високоеластичний стан. Це має місце, коли не встигають відбуватися множинні переміщення центрів тяжіння макромолекул за необоротної деформації. У цих умовах полімер поводиться як квазизшитий, у якому можуть накопичуватия значні оборотні (високоеластичні) деформації, а плинність подавлена. За M/Mкр > 10 перехід полімерів з плинного у високоеластичний стан виражений досить різко. У цьому випадку за швидкостей деформації, які перевищують критичні значення, плинність полімерів виявляється настільки подавленою, що реалізувати сталий ламінарний рух неможливо. Значення Mкр може змінюватися, залежно від природи полімерного ланцюга, в десятки разів. Отже, умови прояву аномалії в’язкості й переходу полімерів з плинного стану у високоеластичний визначає не абсолютне значення молекулярної маси, а відношення M/Mкр.
Переходи з
плинного стану у високоеластичний і
навпаки визначає співвідношення
швидкості деформації і часу релаксації.
Добутком цих показників можна
характеризувати в’язкопружні властивості
полімеру:
θ,
де θ
– час релаксації. Розглянутий вище
перехід полімерів від одного фізичного
стану в інше повинен відбуватися за
певного значення
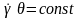 .
Критична швидкість деформації
.
Критична швидкість деформації
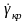 обернено
пропорційна початкової в’язкості
полімеру й, відповідно, залежить від
температури. Але
обернено
пропорційна початкової в’язкості
полімеру й, відповідно, залежить від
температури. Але
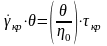 ,
де
,
де
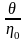 за М >>
Mкр
не повинне залежати від молекулярної
маси й температури, тобто критичне
напруження зсуву τкр
має характерне значення для кожного
полімергомологічного ряду, хоча воно
порівняно слабко залежить від природи
полімерного ланцюга, змінюючись у
крайніх випадках приблизно у 10 разів.
Для карбоцепних полімерів критичне
напруження зсуву підвищується зі
збільшенням кінетичної гнучкості
макромолекул. Найбільш високим критичним
напруженням зсуву відрізняються лінійні
поліолефіни й полібутадієни (τкр
= (3−4)105
Па). Отже, неможливо реалізувати сталу
ламінарну течію високомолекулярних
полімерів за напружень зсуву, що
перевершують наведені значення.
за М >>
Mкр
не повинне залежати від молекулярної
маси й температури, тобто критичне
напруження зсуву τкр
має характерне значення для кожного
полімергомологічного ряду, хоча воно
порівняно слабко залежить від природи
полімерного ланцюга, змінюючись у
крайніх випадках приблизно у 10 разів.
Для карбоцепних полімерів критичне
напруження зсуву підвищується зі
збільшенням кінетичної гнучкості
макромолекул. Найбільш високим критичним
напруженням зсуву відрізняються лінійні
поліолефіни й полібутадієни (τкр
= (3−4)105
Па). Отже, неможливо реалізувати сталу
ламінарну течію високомолекулярних
полімерів за напружень зсуву, що
перевершують наведені значення.
|
Рис. 3.7. Залежність
Q від Δ |
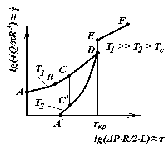
Ділянка АВ відповідає ламінарному плину рідини, що подібна ньютонівській рідині на даній ділянці кривої плину. Починаючи від точки В, для якої τ/τкр ≈ 0,2, спостерігається коливальний режим нестійкго плину, що називається «еластичною турбулентністю» або «дробленням розплаву». Еластична турбулентність проявляється під впливом розвитку великих оборотних деформацій, які на режимах плину, що відповідають ділянці кривій ВСD, можуть досягати багатьох десятків відсотків.
Ділянці плину СD відповідає помітна аномалія в’язкості, що виражена тим слабкіше, чим вужче молекулярно-масовий розподіл полімеру й вище температура плину. Точа D відповідає досягненню критичних значень швидкостей і напружень зсуву, за яких відбувається перехід полімеру у високоеластичний стан. Це супроводжується різким зниженням його плинності й зменшенням адгезії розплаву до стінки капіляра. У результаті спостерігається падіння опору руху полімеру в каналі й, відповідно, – стрибкоподібне підвищення витрати − ефект зриву потоку (вертикальна гілка DЕ). Унаслідок зриву потоку ламінарний плин полімеру перетворюється у ковзання відносно стінок каналу, тому в цьому режимі руху розрахунок швидкості зсуву стає неможливим. У результаті ковзання полімеру в каналі й супутнього йому падіння опору, перепад тиску, що відповідає критичному напруженню зсуву, зосереджується у вхідній зоні каналу, де різко зростає швидкість руху полімеру. Це призводить до розриву безперервності середовища як будь-якого пружньо–напруженого тіла (гілка ЕF). Хоча положення точки Е слабко залежить від молекулярної маси полімеру в межах даного гомологічного ряду, положення гілки АD значною мірою залежить від її, причому критична швидкість зсуву знижується приблизно на порядок у разі підвищення молекулярної маси у два рази.
За значного зниження температури, аномалія в’язкості досить різко виражена в області високих напружень зсуву, що передують зриву потоку. Отже, за низьких температур навіть при незначному розширенні ММР перехід від ньютонівського плину до гілки зриву може бути плавним, а критична точка D неявно вираженою (крива СD).
Уведення в полімер дисперсних наповнювачів спричиняє різке зростання в’язкості, але не змінює її температурного коефіцієнта. Отже, механізм плину наповнених полімерів майже не відрізняється від механізму плину ненаповнених полімерів, тобто підчас плину не відбувається розриву зв’язків між полімером і наповнювачем. Частки наповнювача є центрами суцільної просторової сітки, утворюваної в результаті орієнтації ланцюгів полімеру під впливом силового поля часток наповнювача (якщо наповнювач хімічно активний). Фіксування молекули полімеру на поверхні часток наповнювача передує утворенню навколо частки наповнювача оболонки з полімерних молекул, у яких полімер має підвищені механічні властивості. Для термопластичних полімерів також характерне утворення механічно міцної суцільної просторової структури. Такі структури мають тиксотропні властивості, причому для них характерна наявність тонких залишкових прошарків рідкого середовища в місцях контакту між частками. Ці прошарки, знижуючи міцність системи, забезпечують її здатність до помітних пластичних деформацій (пластичного плину) без значного руйнування структури, і до легкого її відновлення після повного або часткового руйнування. За цим передбачається, що у разі високих ступенів наповнення, внаслідок взаємодії самих часток наповнювача між собою, у системі можуть виникати ланцюжкові й сітчасті структури.