

Гемоглобинопатии
Все структурные аномалии белковой части гемоглобина называют гемоглобинозами. Различают:
-
гемоглобинопатии;
-
талассемии.
Гемоглобинопатии – наследственные изменения структуры какой-либо цепи нормального гемоглобина вследствие точечных мутаций генов. Известно около 300 вариантов HbA, имеющих в первичной структуре α- или β-цепи незначительные изменения. Некоторые из них практически не влияют на функции белка и здоровье человека, другие – вызывают значительные нарушения функции HbA и развитие заболеваний различной степени тяжести.
В аномальных гемоглобинах изменения могут затрагивать аминокислоты:
-
находящиеся на поверхности белка;
-
участвующие в формировании активного центра;
-
аминокислоты, замена которых нарушает трехмерную конформацию молекулы;
-
аминокислоты, замена которых изменяет четвертичную структуру белка и его регуляторные свойства.
Аномальные гемоглобины отличаются от HbA по первичной структуре, форме, величине заряда. При этом изменяются такие свойства как сродство к кислороду, растворимость, устойчивость к денатурации и др.
Примеры.
-
Серповидноклеточная анемия. Наследственное заболевание, связанное с заменой глутаминовой кислоты в 6-м положении (с N-конца) на валин в β-цепях молекулы гемоглобина S. Растворимость дезоксигемоглобина S значительно снижена. Его молекулы начинают «слипаться», образуя волокнистый осадок, который деформирует эритроцит, придавая ему форму серпа (полумесяца). Такие эритроциты плохо проходят через капилляры тканей, закупоривают сосуды и создают локальную гипоксию. Они быстро разрушаются и возникает гемолитическая анемия. Дети, гомозиготные по мутантному гену, часто умирают в раннем возрасте. Болезнь распространена в странах Южной Америки, Африки и Юго-Восточной Азии.
-
Гемоглобин М – в результате мутации в гене происходит замена в α- или β-цепи гистидина (в 7-м или 8-м положении) на тирозин. В результате этого Fe2+ окисляется в Fe3+ и образуется метгемоглобин, не способный связывать кислород. Развивается цианоз и гипоксия тканей.
Талассемии – наследственные заболевания, связанные с нарушением синтеза α- или β-цепей.
β-талассемии развиваются в результате снижения синтеза β-цепей. Проявляется после рождения, при этом в крови наряду с НbА появляется до 15 % НbА2 и 15-60 % HbF. Болезнь характеризуется гиперплазией и разрушением костного мозга, поражением печени, селезенки и сопровождается гемолитической анемией.
α-талассемии возникают при нарушении синтеза α-цепей. При полном отсутствии α-цепей наступает внутриутробная гибель плода, так как не образуется HbF, а тетрамеры γ4 обладают высоким сродством к кислороду и не способны выполнять транспортную функцию, что ведет к развитию тканевой гипоксии и к смерти вскоре после рождения.
Обмен железа
В организме взрослого человека содержится 3-4 г железа, из этого количества около 3,5 г находится в плазме крови. Гемоглобин эритроцитов содержит примерно 68 % всего железа организма, ферритин – 27 % (резервное железо печени, селезенки, костного мозга), миоглобин (в мышцах) – 4 %, трансферрин (в плазме крови) – 0,1. На долю всех содержащих железо ферментов приходится примерно 1 % железа, имеющегося в организме.
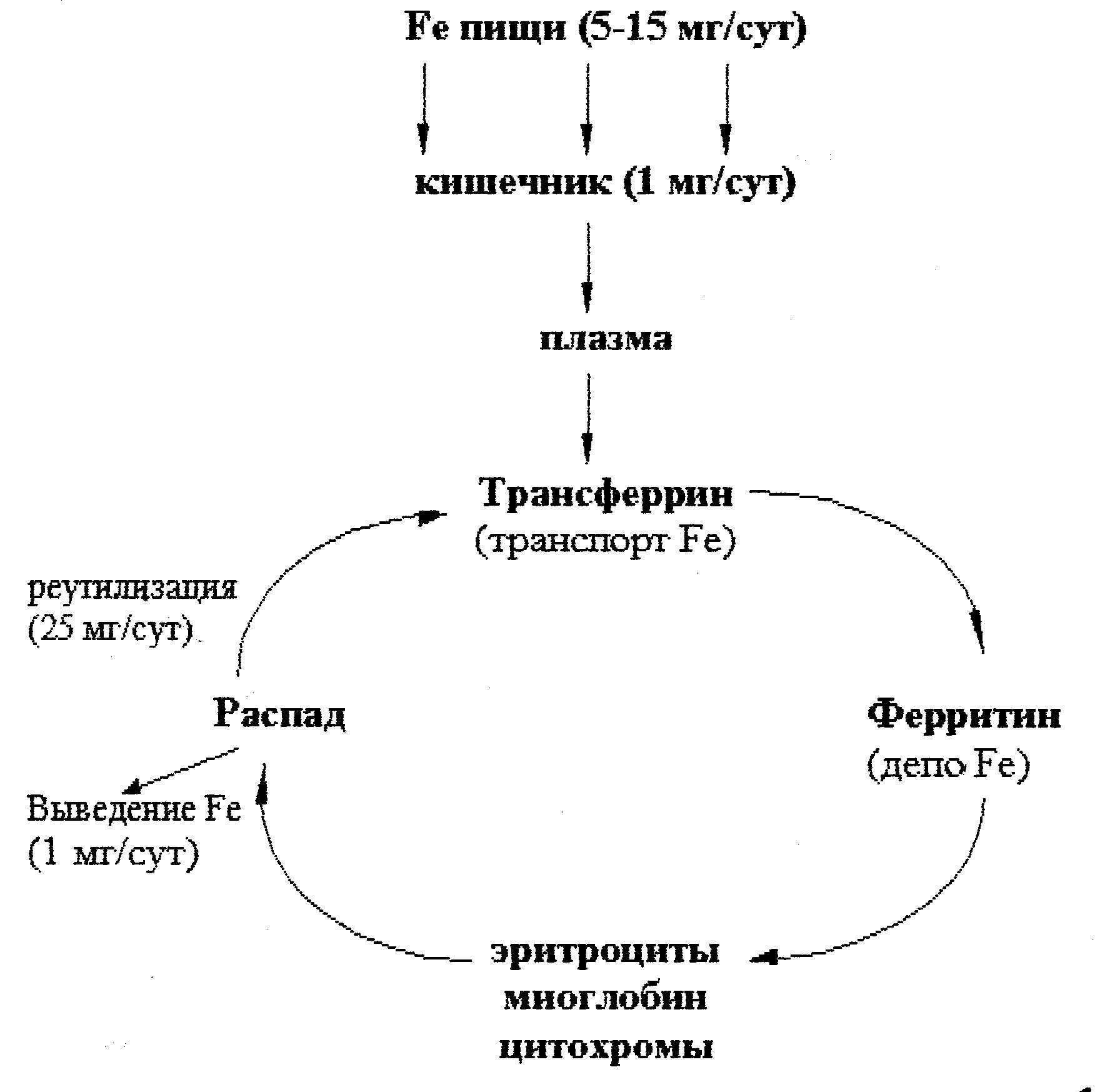
Рис. 30.1. Обмен железа в организме человека.
В обмене железа принимает участие ряд белков.
Апоферритин. Белок связывает железо в эритроцитах и превращается в ферритин, который остается в энтероцитах. Таким способом регулируется поступление железа в капилляры крови из клеток кишечника. Когда потребность организма в железе невелика, скорость синтеза апоферритина повышается. При недостатке железа в организме апоферритин в энтероцитах почти не синтезируется.
Трансферрин. Это транспортный белок, относится к гликопротеинам, синтезируется в печени. Он имеет два центра связывания железа. Трансферрин транспортирует железо с током крови к местам депонирования и использования. В норме трансферрин насыщен железом приблизительно на 33 %.
Ферритин. Олигомерный белок с молекулярной массой 450 к Да. Он состоит из 24 идентичных протомеров, образующих полую сферу. Железо депонируется в ферритине в виде гидроксифосфата. Содержание железа в молекуле ферритина непостоянно. Функция ферритина – депонирование железа. Ферритин содержится почти во всех тканях, но в наибольшем количестве в печени, селезенке, костном мозге.