

Учебники / Genetics and Auditory Disorders Keats 2002
.pdfKallmann |
XL (AD) |
KAL1/ |
Neural cell |
Axonal path- |
Occas mild |
Hypogonadism; anosmia, |
|
Bick et al. 1992; |
syndrome |
(AR); |
HHA/ |
adhesion |
finding? |
SNHL or |
agenesis of olfactory |
|
Legouis et al. 1991 |
|
Xp22.3 |
KALIG-1/ |
molecule |
|
mod-severe |
lobes |
|
|
|
|
ADMLX |
|
|
MHL |
|
|
|
Kniest dysplasia |
Sporadic, |
COL2A1 |
Collagen a1(II) |
Fibrillar |
CHL, SNHL, |
Skeletal abnormalities; |
COL2A1, |
Garofalo et al. 1991; |
(metatropic |
AD; |
|
|
collagen- |
or MHL |
cleft palate |
Col2a1 |
Vandenberg et al. |
dysplasia, Type II) |
12q13.11- |
|
|
cartilage |
|
|
transgenic |
1991; Winterpacht |
|
q13.2 |
|
|
|
|
|
knockins; |
et al. 1993; Li et al. |
|
|
|
|
|
|
|
Col2a1 +/- |
1995 |
|
|
|
|
|
|
|
heterozygous |
|
|
|
|
|
|
|
|
knockout |
|
Krabbe disease |
AR; |
GALC/ |
Galactosylcera- |
Lysosomal |
Progressive |
Central nervous system |
Twitcher, twi |
Sakai et al. 1996; |
|
14q24.3- |
GLD/ |
mide beta- |
enzyme |
SNHL |
degeneration; progresive |
|
Wenger et al. |
|
q32.1 |
GCL |
galactosidase |
|
|
blindness |
|
1997 |
Marfan syndrome |
AD; |
MFS1/ |
Fibrillin-1 |
Formation of |
CHL or |
Skeletal, ocular, |
|
Dietz et al. 1991 |
|
15q21.1 |
MFS/ |
|
microfibrils |
SNHL |
cardiovascular anomalies |
|
|
|
|
FBN1 |
|
|
|
|
|
|
|
3p24.2-p25 |
MFS2 |
Unknown |
Unknown |
|
Skeletal, cardiovascular |
|
Collod et al. 1994 |
|
|
|
|
|
|
anomalies |
|
|
Marshall syndrome |
AD; |
COL11A1 |
Collagen |
Fibrillar |
Progressive |
Skeletal, joint |
Chondrodysplasia, |
Li et al. 1995; Griffith |
|
1p21 |
|
a1(XI) |
collagen- |
SNHL |
abnormalities; myopia; |
cho |
et al. 1998; Annunen |
|
|
|
|
cartilage |
|
cataracts; craniofacial |
|
et al. 1999 |
|
|
|
|
|
|
dysmorphism |
|
|
Mohr-Tranebjaerg |
XL; |
MTS/ |
Unknown |
Unknown |
Progressive |
Blindness; dystonia; |
|
Jin et al. 1996 |
syndrome |
Xq22 |
DDP/ |
|
|
SNHL |
mental deficiency; |
|
|
|
|
DFN1 |
|
|
|
fractures |
|
|
Multiple synostoses |
AD; |
SYNS1 |
Unknown |
Unknown |
Progressive |
Premature joint fusions; |
|
Krakow et al. 1998 |
syndrome 1 |
17q21-q22 |
|
|
|
CHL |
skeletal abnormalities |
|
|
Neurofibromatosis, |
AD; |
NF2/ |
MERLIN/ |
Tumor |
Progressive |
Schwannomas of other |
|
Rouleau et al. 1993; |
Type 2 |
22q12.2 |
BANF/ |
Schwannomin |
suppressor |
SNHL; |
nerves; brain tumors; |
|
Trofatter et al. 1993; |
|
|
CAN/ |
|
|
vestibular |
cataracts; café-au-lait |
|
Trofatter et al. 1993 |
|
|
SCH |
|
|
dysfunction |
spots; subcutaneous |
|
|
|
|
|
|
|
|
neurofibromas |
|
|
171 Disorders Auditory Linked-X and Autosomal .6

TABLE 6.5. Continued
|
Inheritance |
Locus |
|
Gene |
Auditory |
|
Mouse |
Selected |
Syndrome |
and Location |
Symbol |
Gene Product |
Function |
Phenotype |
Associated Pathology |
Model |
References |
Niemann-Pick |
AR; |
NPC/ |
Similarity to |
Regulation |
Progressive |
Progressive neurologic |
Sphingomyelinosis, |
Carstea et al. 1997; |
disease, Type C |
18q11-q12 |
NPC1 |
transmembrane |
of intra- |
SNHL |
deterioration due to |
spm |
Loftus et al. 1997 |
|
|
|
domains, |
cellular |
|
sphingomyelin |
|
|
|
|
|
cholesterol- |
cholesterol |
|
accumulation |
|
|
|
|
|
sensing regions |
trafficking |
|
|
|
|
Noonan syndrome |
AD; |
NS1 |
Unknown |
Unknown |
Progressive |
Skeletal, craniofacial, |
|
Jamieson et al. 1994 |
|
12q24 |
|
|
|
SNHL or |
heart anomalies; mild |
|
|
|
|
|
|
|
MHL |
mental retardation; |
|
|
|
|
|
|
|
|
hematologic |
|
|
|
|
|
|
|
|
abnormalities; |
|
|
|
|
|
|
|
|
lymphangiomas, |
|
|
|
|
|
|
|
|
schwannomas |
|
|
Norrie disease |
XLR; |
ND/NDP |
Homology to |
role in |
Progressive |
Congenital or progressive |
NDP -/- |
Berger et al. 1992; |
|
Xp11.4 |
|
mucins |
neuroectoder- |
SNHL |
blindness; mental |
knock-out |
Chen et al. 1992; |
|
|
|
|
mal cell-cell |
(cochlear) |
deficiency |
|
Meindl et al. 1992; |
|
|
|
|
interactions? |
|
|
|
Berger et al. 1996 |
Ocular albinism |
Xp22.3 |
OASD |
Unknown |
Unknown |
Late-onset |
Ocular albinism |
|
Winship et al. |
with sensorineural |
|
|
|
|
progressive |
|
|
1993 |
deafness |
|
|
|
|
SNHL |
|
|
|
Orofaciodigital |
XLD; |
OFD1 |
Unknown |
Unknown |
Occas CHL |
Midfacial clefting; |
X-linked |
Sweet and Lane 1980; |
syndrome, Type 1 |
Xp22.3- |
|
|
|
|
hyperplasia of oral cavity |
polydactyly, |
Feather et al. 1997 |
|
p22.2 |
|
|
|
|
frenula; cleft tongue; |
Xpl |
|
|
|
|
|
|
|
hand anomalies; |
|
|
|
|
|
|
|
|
polycystic kidneys |
|
|
Osteogenesis |
AD (AR); |
OI/ |
Collagen a1(I) |
Fibrillar |
Progressive |
Brittle and deformed |
Mov-13, |
Bonadio et al. 1990; |
imperfecta,Type I, |
17q21.31- |
COL1A1 |
|
collagen- |
CHL or |
bones, hyperextensible |
retroviral |
Byers 1993; Pereira |
Type II, Type III, |
q22.05 |
|
|
bone, |
MHL |
joints; blue sclerae |
insertion into |
et al. 1993 |
Type IV |
|
|
|
tendon, skin |
|
|
col1a1; |
|
|
|
|
|
|
|
|
Transgenic |
|
internal deletion of
COL1A1
Friedman .B.T and Griffith .J.A 172
|
7q22.1 |
OI/ |
Collagen a2(I) |
Fibrillar |
Same as |
Same as above |
|
Byers 1993 |
|
|
COL1A2 |
|
collagen- |
above |
|
|
|
|
|
|
|
bone, |
|
|
|
|
|
|
|
|
tendon, skin |
|
|
|
|
Osteopetrosis |
AR; |
OPTB1 |
Unknown |
Unknown |
MHL or |
Facial palsy; visual loss; |
Osteosclerosis, |
Heaney et al. 1998 |
(Albers-Schönberg |
11q12-q13 |
|
|
|
CHL |
generalized osteosclerosis |
oc |
|
disease) |
|
|
|
|
|
|
|
|
Type II |
AD; |
OPTA2 |
Unknown |
Unknown |
CHL |
Facial palsy; generalized |
|
Van Hul et al. 1997 |
|
1p21 |
|
|
|
|
osteosclerosis |
|
|
Otopalatodigital |
XL; |
OPD1 |
Unknown |
Unknown |
CHL |
Craniofacial, skeletal |
|
Hoar et al. 1992 |
syndrome, Type I |
Xq28 |
|
|
|
|
anomalies |
|
|
Otospondylomega- |
AR, AD; |
OSMED/ |
Collagen |
Fibrillar |
Mod-severe |
Skeletal and craniofacial |
|
Vikkula et al. 1995; |
epiphyseal |
6p21.3 |
WZS/ |
a2(XI) |
collagen- |
SNHL |
abnormalities; myopia |
|
Pihlajamaa et al. |
dysplasia |
|
COL11A2 |
|
cartilage |
|
|
|
1998; |
Paget disease |
AD; |
PDB1 |
Unknown |
Unknown |
CHL, MHL, |
Progressive skull |
|
Fotino et al. |
|
6p21.3 |
|
|
|
or SNHL |
enlargement; bending of |
|
1977 |
|
|
|
|
|
(cochlear); |
weight-bearing bones; |
|
|
|
|
|
|
|
vestibular |
neurologic deficits |
|
|
|
|
|
|
|
dysfunction |
|
|
|
|
18q21-q22 |
PDB2 |
Unknown |
Unknown |
|
|
|
Cody et al. 1997 |
Pendred syndrome |
AR; |
PDS |
Similarity to |
Chloride/ |
Congenital |
Thyroid organification |
|
Everett et al. 1997; |
|
7q31 |
|
transmembrane |
iodine |
SNHL |
defect, goiter |
|
Coyle et al. 1998; |
|
|
|
sulfate |
transporter? |
(cochlear); |
|
|
Scott et al. 1998 |
|
|
|
transporters |
|
vestibular |
|
|
|
|
|
|
|
|
dysfunction |
|
|
|
Pfeiffer syndrome |
AD; |
ACS5/ |
Fibroblast |
Tyrosine |
CHL |
Craniosynostosis; digit |
|
Muenke et al. 1994 |
|
8p11.2-p11.1 |
FGFR1 |
growth factor |
kinase |
|
abnormalities |
|
|
|
|
|
receptor 1 |
growth |
|
|
|
|
|
|
|
|
factor |
|
|
|
|
|
|
|
|
receptor |
|
|
|
|
|
10q26 |
ACS5/ |
Fibroblast |
Tyrosine |
CHL |
Same as above |
|
Lajeunie et al. 1995; |
|
|
FGFR2 |
growth factor |
kinase |
|
|
|
Rutland et al. 1995 |
|
|
|
receptor 2 |
growth |
|
|
|
|
|
|
|
|
factor |
|
|
|
|
|
|
|
|
receptor |
|
|
|
|
173 Disorders Auditory Linked-X and Autosomal .6

TABLE 6.5. Continued
|
Inheritance |
Locus |
|
Gene |
Auditory |
|
Mouse |
Selected |
Syndrome |
and Location |
Symbol |
Gene Product |
Function |
Phenotype |
Associated Pathology |
Model |
References |
|
4p16.3 |
ACS5/ |
Fibroblast |
Tyrosine |
CHL |
Same as above |
|
Bellus et al. 1996 |
|
|
FGFR 3 |
growth factor |
kinase |
|
|
|
|
|
|
|
receptor 3 |
growth |
|
|
|
|
|
|
|
|
factor |
|
|
|
|
|
|
|
|
receptor |
|
|
|
|
Piebaldism |
AD; |
PBT/KIT |
KIT |
Mast/stem |
Progressive |
Pigmentation |
Dominant |
Geissler et al. 1988; |
|
4q11-q12 |
|
protooncogene |
cell growth |
SNHL |
abnormalities; ataxia; |
white |
Giebel and Spritz |
|
|
|
|
factor |
|
mental retardation |
spotting, W |
1991; Spritz and |
|
|
|
|
|
|
|
|
Beighton 1998 |
Refsum disease |
AR; |
HMSN IV/ |
Phytanoyl-CoA |
Peroxisomal |
Progressive |
Retinitis pigmentosa |
|
Jansen et al. |
|
10pter-p11.2 |
PAHX/ |
hydroxylase |
enzyme |
SNHL |
(retinal degeneration, |
|
1997; Mihalik et al. |
|
|
PHYH |
|
|
|
blindness); cerebellar |
|
1997 |
|
|
|
|
|
|
ataxia; increased plasma |
|
|
|
|
|
|
|
|
phytanic acid |
|
|
Refsum disease, |
AR; |
IRD/ |
Peroxisome |
Peroxisomal |
Prof SNHL |
Retinitis pigmentosa; |
|
Reuber et al. 1997 |
infantile form |
7q21-q22 |
PEX1 |
biogenesis |
matrix |
|
mental retardation; |
|
|
|
|
|
factor 1 |
protein |
|
craniofacial dysmorphism; |
|
|
|
|
|
|
import |
|
liver dysfunction; short |
|
|
|
|
|
|
|
|
stature |
|
|
Renal tubular |
AR; |
dRTA/ |
B1-subunit of |
Proton pump |
Progressive |
Impaired renal tubular |
|
Karet et al. 1999 |
acidosis with |
2cen-q13 |
ATP6B1 |
H+-ATPase |
|
SNHL |
acid secretion |
|
|
sensorineural |
|
|
|
|
|
|
|
|
deafness |
|
|
|
|
|
|
|
|
Saethre-Chotzen |
AD; |
SCS/ |
TWIST |
Transcription |
Occas CHL |
Premature fusion of |
Twist +/- |
el Ghouzzi et al. |
syndrome |
7p21 |
ACS3/ |
|
factor |
or MHL |
cranial sutures; digit |
heterozygous |
1997; Howard et al. |
|
|
TWIST |
|
|
|
abnormalities |
knockout |
1997; Bourgeois et al. |
|
10q26 |
SCS/ |
Fibroblast |
Tyrosine |
Occas CHL |
Same as above |
Fgfr2 -/- |
1998 Arman et al. |
|
|
ACS3/ |
growth factor |
kinase |
or MHL |
|
knockout |
1998; Paznekas et al. |
|
|
FGFR2 |
receptor 2 |
growth |
|
|
|
1998 |
|
|
|
|
factor |
|
|
|
|
|
|
|
|
receptor |
|
|
|
|
Friedman .B.T and Griffith .J.A 174
|
4p16.3 |
SCS/ |
Fibroblast |
Tyrosine |
Occas CHL |
Same as above |
Fgfr3 -/- |
|
|
ACS3/ |
growth factor |
kinase |
or MHL |
|
knockout |
|
|
FGFR3 |
receptor 3 |
growth |
|
|
|
|
|
|
|
factor |
|
|
|
|
|
|
|
receptor |
|
|
|
Sialidosis |
AR; |
NEU/ |
Sialidase/ |
Lysosomal |
CHL or |
Central nervous system |
SM/J line |
|
6p21.3 |
NEU1 |
Neuraminidase |
enzyme |
MHL |
degeneration; vision loss; |
|
|
|
|
|
|
|
dysostosis; facial |
|
|
|
|
|
|
|
dysmorphism |
|
Smith-Magenis |
Sporadic; |
SMS/ |
Contiguous |
Multiple |
CHL, occas |
Somatic, mental |
|
syndrome |
17p11.2 |
SMCR |
gene deletion |
deleted genes |
SNHL |
retardation; behavioral |
|
|
|
|
including |
|
|
abnormalities; nonspecific |
|
|
|
|
MYO15 |
|
|
combinations of |
|
|
|
|
|
|
|
anomalies |
|
Spondyloepiphyseal |
AD; |
SEDC/ |
Collagen al (II) |
Fibrillar |
Occas. mod- |
Skeletal abnormalities; |
|
dysplasia congenita |
12q13.11- |
COL2A1 |
|
collagen- |
severe high- |
cleft palate; short stature |
|
|
q13.2 |
|
|
cartilage |
freq. SNHL |
|
|
Stickler syndrome, |
AD; |
STL1/ |
Collagen a1(II) |
Fibrillar |
Progressive |
Skeletal and joint |
COL2A1, |
Type I |
12q13.11- |
COL2A1 |
|
collagen- |
SNHL, occas |
abnormalities; |
Col2a1 |
|
q13.2 |
|
|
cartilage |
CHL |
myopia; cataracts; |
transgenic |
|
|
|
|
|
|
craniofacial dysmorphism |
knockins; |
|
|
|
|
|
|
|
Col2a1 +/- |
|
|
|
|
|
|
|
heterozygous |
|
|
|
|
|
|
|
knockout |
Type II |
AD; |
STL2/ |
Collagen |
Fibrillar |
SNHL |
Same as type I Stickler’s |
|
|
6p21.3 |
COL11A2 |
a2(XI) |
collagen- |
|
syndrome, but no ocular |
|
|
|
|
|
cartilage |
|
manifestations |
|
Type III |
AD; |
STL3/ |
Collagen |
Fibrillar |
SNHL |
Same as type I Stickler’s |
Chondrodys- |
|
1p21 |
COL11A1 |
a1(XI) |
collagen- |
|
syndrome |
plasia, cho |
|
|
|
|
cartilage |
|
|
|
Symphalangism, |
AD; |
SYM1 |
Unknown |
Unknown |
CHL |
Fusion of extremity joints |
|
proximal |
17q21-q22 |
|
|
|
|
|
|
Tay-Sachs disease |
AR; |
TSD/ |
Hexosaminidase |
Lysosomal |
SNHL |
Progressive mental, motor |
Hexa -/- |
|
15q23-q24 |
HEXA |
A |
enzyme |
|
retardation; seizures; |
knockout |
|
|
|
|
|
|
blindness |
|
Colvin et al. 1996; Deng et al. 1996; Paznekas et al. 1998
Bonten et al. 1996; Pshezhetsky et al. 1997; Rottier et al. 1998
Chen et al. 1997; Smith et al. 1986
Garofalo et al. 1991; Vandenberg et al. 1991; Spranger et al. 1994; Li et
al. 1995
Vikkula et al. 1995; Sirko-Osadsa et al. 1998
Li et al. 1995; Richards et al. 1996; Annunen et al. 1999
Polymeropoulos et al. 1995
Sango et al. 1995; Myerowitz 1997
175 Disorders Auditory Linked-X and Autosomal .6

TABLE 6.5. Continued
|
Inheritance |
Locus |
|
Gene |
Auditory |
|
Mouse |
Selected |
Syndrome |
and Location |
Symbol |
Gene Product |
Function |
Phenotype |
Associated Pathology |
Model |
References |
Tietz syndrome |
AD; |
MITF |
Microphthalmia |
Transcription |
Congenital |
Skin/hair albinism |
microphthal- |
Steingrimsson et al. |
|
3p14.1-p12.3 |
|
-associated |
factor |
prof SNHL; |
|
mia, mi |
1994; Amiel et al. |
|
|
|
transcription |
|
normal |
|
|
1998; Smith et al. |
|
|
|
factor |
|
vestibular |
|
|
1997 |
|
|
|
|
|
function |
|
|
|
Townes-Brocks |
AD; |
TBS/ |
C2H2 zinc |
Transcription |
SNHL |
Deformities of external |
|
Kohlhase et al. 1998 |
syndrome |
16q12.1 |
SALL1 |
finger |
factor |
|
ears, anus, digits, kidneys, |
|
|
|
|
|
transcription |
|
|
and heart |
|
|
|
|
|
factor |
|
|
|
|
|
Treacher Collins’ |
AD; |
TCOF1/ |
Nucleolar |
Nucleolar |
Variable |
Craniofacial anomalies; |
|
Treacher Collins, |
syndrome |
5q32-q33.1 |
TCS/ |
phosphoprotein |
protein |
CHL |
eyelid colobomas |
|
syndrome |
|
|
MFD1 |
|
trafficking? |
|
|
|
collaborative group |
|
|
|
|
|
|
|
|
1996; Wise et al. 1997 |
Usher syndrome, |
AR; |
USH1A/ |
Unknown |
Unknown |
Congenital |
Onset of retinitis |
|
Kaplan et al. 1992 |
Type 1A |
14q32 |
USH1 |
|
|
severe-prof |
pigmentosa (retinal |
|
|
|
|
|
|
|
SNHL; |
degeneration, blindness) |
|
|
|
|
|
|
|
absent |
by 10 yrs |
|
|
|
|
|
|
|
vestibular |
|
|
|
|
|
|
|
|
function |
|
|
|
Type 1B |
11q13.5 |
USH1B/ |
Type VIIA |
Intracellular |
Same as |
Same as above |
shaker-1, sh1 |
Gibson et al. 1995; |
|
|
MYO7A |
myosin- |
actin-based |
above |
|
|
Weil et al. 1995 |
|
|
|
unconventional |
transport? |
|
|
|
|
Type 1C |
11p15.1 |
USH1C |
Unknown |
Unknown |
Same as |
Same as above |
|
Keats et al. 1994 |
|
|
|
|
|
above |
|
|
|
Type 1D |
10q |
USH1D |
Unknown |
Unknown |
Same as |
Same as above |
|
Wayne et al. 1996 |
|
|
|
|
|
above |
|
|
|
Type 1E |
21q21 |
USH1E |
Unknown |
Unknown |
Same as |
Same as above |
|
Chaib et al. 1997 |
|
|
|
|
|
above |
|
|
|
Type 1F |
10 |
USH1F |
Unknown |
Unknown |
Same as |
Same as above |
|
Wayne et al. 1997 |
|
|
|
|
|
above |
|
|
|
Friedman .B.T and Griffith .J.A 176
Type 2A |
AR; |
USH2A |
Contains |
Extracellular |
Congenital |
Onset of retinitis |
|
Eudy et al. 1998 |
|
1q41 |
|
laminin-EGF |
matrix/ |
mod-severe |
pigmentosa in late |
|
|
|
|
|
and fibronectin |
adhesion |
SNHL; |
teens/early adulthood |
|
|
|
|
|
domains |
molecule? |
normal |
|
|
|
|
|
|
|
|
vestibular |
|
|
|
|
|
|
|
|
function |
|
|
|
Type 2B |
5q14.3-q21.3 |
USH2B |
Unknown |
Unknown |
Same as |
Same as above |
|
Pieke-Dahl et al. 1993; |
|
|
|
|
|
above |
|
|
Pieke-Dahl et al. 1998 |
Type 3 |
AR; |
USH3 |
Unknown |
Unknown |
Progressive |
Variable onset of retinitis |
|
Sankila et al. 1995 |
|
3q21-q25 |
|
|
|
SNHL; |
pigmentosa |
|
|
|
|
|
|
|
normal or |
|
|
|
|
|
|
|
|
decreased |
|
|
|
|
|
|
|
|
vestibular |
|
|
|
|
|
|
|
|
function |
|
|
|
Van Buchem disease AR; |
VBCH |
Unknown |
Unknown |
MHL or |
Skeletal hyperostosis |
|
Van Hul et al. 1998 |
|
|
17q11.2 |
|
|
|
SNHL |
|
|
|
Velocardiofacial |
AD; |
VCFS |
Frequent |
Multiple |
CHL (assoc |
Heart anomalies; facial |
|
Driscoll et al. 1992 |
(Shprintzen) |
22q11 |
|
contiguous gene |
deleted |
w/otitis |
dysmorphism; palatal |
|
|
syndrome |
|
|
deletion |
genes |
media), |
cleft/dysfunction; mild |
|
|
|
|
|
|
|
occas SNHL |
mental retardation |
|
|
Vohwinkel |
13q12 |
KHM/ |
Gap junction |
Gap junction |
SNHL |
Mutilating keratoderma |
|
Maestrini et al. 1999 |
syndrome, classic |
|
GJB2/ |
beta-2 subunit |
subunit |
|
|
|
|
form |
|
CX26 |
|
|
|
|
|
|
Vohwinkel |
AD; |
LOR |
loricrin |
Structural |
Congenital |
Hyperkeratosis and other |
|
Maestrini et al. 1996 |
syndrome, variant |
1q21 |
|
|
component |
and/or |
skin anomalies |
|
|
form |
|
|
|
of cell |
progressive |
|
|
|
|
|
|
|
envelope of |
SNHL |
|
|
|
|
|
|
|
epidermis |
|
|
|
|
Waardenburg |
AD; |
WS1/ |
Paired-box |
Transcription |
Occas |
Craniofacial dysmorphism, |
splotch, sp |
Epstein, Vekemans, |
syndrome, Type I |
2q35 |
PAX3 |
DNA-binding |
factor |
congenital, |
including dystopia |
|
and Gros 1991; |
|
|
|
protein |
|
variable |
canthorum; pigmentation |
|
Baldwin et al. 1992; |
|
|
|
|
|
SNHL |
abnormalities |
|
Tassabehji et al. 1992 |
Type II |
AD; |
WS2/ |
Microphthalmia |
Transcription |
Same as |
Craniofacial dysmorphism |
microphthal- |
Steingrimsson et al. |
|
3p14.1-p12.3 |
WS2A/ |
-associated |
factor |
WS1, SNHL |
without dystopia |
mia, mi |
1994; Tassabehji, |
|
|
MITF |
transcription |
|
may be |
canthorum; pigmentation |
|
Newton, and Read |
|
|
|
factor |
|
progressive |
abnormalities |
|
1994 |
177 Disorders Auditory Linked-X and Autosomal .6

TABLE 6.5. Continued
|
Inheritance |
Locus |
|
Gene |
Auditory |
|
Mouse |
Selected |
Syndrome |
and Location |
Symbol |
Gene Product |
Function |
Phenotype |
Associated Pathology |
Model |
References |
Type II, |
Autosomal |
WS2-OA/ |
Microphthalmia |
Transcription |
Progressive |
Ocular albinism |
|
Bard, 1978; Morell |
with ocular |
digenic; |
MITF |
-associated |
factor |
SNHL |
|
|
et al. 1997 |
albinism |
3p14.1- |
|
transcription |
|
|
|
|
|
|
p12.3 |
|
factor |
|
|
|
|
|
|
11q14-q21 |
OCA1/ |
Tyrosinase |
Tyrosinase |
|
|
|
|
|
|
TYR |
|
|
|
|
|
|
Type III (Klein- |
AD, AR; |
WS3/ |
Paired-box |
Transcription |
SNHL |
Same as WS1, with |
splotch, sp |
Epstein et al. |
Waardenburg) |
2q35 |
PAX3 |
DNA-binding |
factor |
|
skeletal abnormalties |
|
1991; Hoth et al. |
|
|
|
protein |
|
|
|
|
1993; Zlotogora et al. |
|
|
|
|
|
|
|
|
1995 |
Type IV (Shah- |
AR; |
WS4/ |
Endothelin-3 |
Extracellular |
SNHL |
Same as WS2, with |
Spotting |
Baynash et al. 1994; |
Waardenburg) |
20q13.2- |
EDN3 |
|
signalling |
|
Hirschsprung disease |
lethal, sl; |
Edery et al. 1996; |
|
q13.3 |
|
|
peptide |
|
(lack of autonomic |
edn3 -/- |
Hofstra et al. 1996 |
|
|
|
|
|
|
innervation to colon) |
knock-out |
|
|
AR; 13q22 |
WS4/ |
Endothelin |
G |
SNHL |
Same as above |
ednrb -/- |
Hosoda et al. 1994; |
|
|
EDNRB |
receptor, type B |
proteincoupled |
|
|
knock-out |
Puffenberger et al. |
|
|
|
|
receptor |
|
|
|
1994; Attie et al. 1995 |
|
AD; 22q13 |
WS4/ |
SRY-related |
Transcription |
SNHL |
Same as above |
Dominant |
Herbarth et al. 1998; |
|
|
SOX10 |
transcription |
factor |
|
|
megacolon, |
Pingault et al. 1998; |
|
|
|
factor |
|
|
|
Dom |
Southard-Smith et al. |
|
|
|
|
|
|
|
|
1998 |
Wolfram syndrome |
AR; |
WFS/ |
Wolframin |
Unknown |
Progressive |
Progressive blindness, |
|
Inoue et al. 1998; |
|
4p16.1 |
WFS1c |
|
|
SNHL, |
diabetes mellitus, diabetes |
|
Strom et al. 1998 |
|
|
|
|
|
HF > LF |
insipidus |
|
|
Xeroderma |
AR; |
XPA/ |
Zinc finger |
DNA |
Progressive |
Photosensitivity; |
Xpa -/- |
Tanaka et al. 1990; de |
pigmentosum, |
9q22.3-q31 |
XP1/ |
domain protein |
excision |
SNHL, |
cutaneous malignancies; |
knock-out |
Vries et al. 1995; |
group A |
|
XPAC |
|
repair |
HF > LF |
neurologic abnormalities |
|
Nakane et al. 1995 |
|
|
|
|
|
|
|
|
|
Friedman .B.T and Griffith .J.A 178
6. Autosomal and X-Linked Auditory Disorders |
179 |
A possible pitfall to this line of associative reasoning is that a syndrome may be caused by a contiguous gene deletion, or by linked mutations in separate genes. In this circumstance, a hearing loss gene may be altered in association with a separate gene(s) causing pathology in another organ system. This was observed in a kindred originally used to identify DFNB1 (Kelsell et al. 1997) in which autosomal dominant palmoplantar keratoderma (PPK) was also segregating. Clinical ascertainment of that pedigree identified 3 individuals with PPK but without hearing loss, and linkage of
GJB2 was demonstrated with the hearing loss phenotype but not the PPK phenotype. This kindred illustrates how contiguous gene deletions or co-segregation of mutations have the potential to confound the linkage analysis and any conclusions regarding shared pathogenetic mechanisms between the affected organ systems.
Approximately 100 genes for syndromic hearing loss have now been mapped, and over 60 of these have been identified (Table 6.5). Some of these genes and their corresponding mutations have provided interesting and novel insights into the development and function of the auditory system. The following review will focus on six forms of syndromic hearing loss in which at least some of the causative genes have been identified, and the resulting molecular data has raised and/or answered important questions regarding the molecular basis of auditory function and hearing impairment. A discussion of all forms of syndromic hearing loss (Gorlin et al. 1995) in which the genes have been mapped or identified would require its own volume. Table 6.5 summarizes some of the essential features of approximately 100 forms of syndromic hearing loss.
It is clinically important to identify the cause of hearing loss in families where in auditory dysfunction is accompanied by other serious problems, such as heart conduction problems (Jervell and Lange-Nielsen syndrome), progressive nephritis (Alport syndrome), or progressive loss of vision
(Usher syndrome). Within the congenitally hearing impaired population, the incidences of these three syndromes are estimated to be 0.25%, 1.0%, and 3 to 8%, respectively (Gorlin et al. 1995; Vernon 1959). Hearing loss may be detected before manifestation of other organ system pathologies in all three of these syndromes. For example, the hearing loss in Jervell and Lange–Nielsen syndrome may be evident before the onset of fainting attacks or detection of a cardiac arrhythmia. Life-saving anti-arrhythmic therapy may thus be initiated prophylactically (Ackerman 1998).
5.1 Stickler Syndrome
5.1.1 Phenotype
Stickler syndrome (OMIM 108300, 184840) is an autosomal-dominant disorder characterized by progressive sensorineural hearing loss, skeletal dysplasia, craniofacial dysmorphism, cataracts, and myopia. Marshall syndrome
180 A.J. Griffith and T.B. Friedman
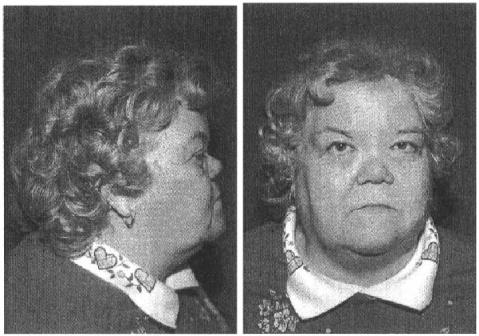
(OMIM 154780) has a similar phenotype, but may be distinguished by a more severe degree of hearing loss and its unique pattern of craniofacial dysmorphism persisting into adulthood (Annunen et al. 1999; Ayme and Preus 1984; Marshall 1958) (Fig. 6.5). The sensorineural hearing loss in these disorders begins during early childhood and progresses over the ensuing decades. The hearing loss is occasionally mixed (Lucarini et al. 1987) and may be due to otitis media or its sequelae, especially in affected children.
Given the characteristic skeletal abnormalities, it is reasonable to postulate that the sensorineural hearing loss might be associated with gross morphogenetic abnormalities of the inner ear. However, there are no reports of temporal bone histopathology for these disorders, although computed tomography (CT) scans of temporal bones of 19 Stickler’s syndrome patients and three Marshall syndrome patients have revealed no abnormalities (Griffith et al. 2000b; Szymko et al. 2000). Therefore, the bony anatomy of the inner ear is at least grossly normal in these disorders.
FIGURE 6.5. Frontal and side views of an individual affected with Marshall syndrome (Griffith et al. 1998). These facial features persisting into adulthood are characteristic of Marshall syndrome and include severe midfacial growth deficiency, a small upturned nose, and a prominent forehead. (Reprinted from Griffith et al.,The American Journal of Human Genetics, Copyright 1998, with permission of The University of Chicago Press.)