

Книги фарма 2 / Bertram G. Katzung-Basic & Clinical Pharmacology(9th Edition)
.pdfLuteinizing hormone is a glycoprotein hormone consisting of two chains and, like FSH, is produced by gonadotroph cells in the anterior pituitary. LH is primarily responsible for regulation of gonadal steroid hormone production. In men, LH acts on testicular Leydig cells to stimulate testosterone production. In the ovary, LH acts in concert with FSH to stimulate follicular development. LH acts on the mature follicle to induce ovulation, and it stimulates the corpus luteum in the luteal phase of the menstrual cycle to produce progesterone and androgens.
There is no LH preparation presently available for clinical use. Human chorionic gonadotropin— with an almost identical structure—is available and can be used as a luteinizing hormone substitute.
Human chorionic gonadotropin is a hormone produced by the human placenta and excreted into the urine, whence it can be extracted and purified. Human chorionic gonadotropin is a glycoprotein consisting of a 92-amino-acid alpha chain virtually identical to that of FSH, LH, and TSH and a beta chain of 145 amino acids that resembles that of LH except for the presence of a carboxyl terminal sequence of 30 amino acids not present in LH.
The function of hCG is to stimulate the ovarian corpus luteum to produce progesterone and maintain the placenta. It is very similar to LH in structure and is used to treat both men and women with LH deficiency.
Pharmacokinetics
Human chorionic gonadotropin is well absorbed after intramuscular administration and has a biologic half-life of 8 hours, compared with 30 minutes for LH. The difference may lie in the high sialic acid content of hCG compared with that of LH. It is apparently modified in the body prior to urinary excretion, because the half-life measured by immunoassay far exceeds that measured by bioassay.
Pharmacodynamics
Human chorionic gonadotropin stimulates production of gonadal steroid hormones. The interstitial and corpus luteal cells of the female produce progesterone, and the Leydig cells of the male produce testosterone. hCG can be used to mimic a midcycle LH surge and trigger ovulation in a hypogonadotropic woman.
Clinical Pharmacology
Diagnostic Uses
In prepubertal boys with undescended gonads, hCG can be used to distinguish a truly retained (cryptorchid) testis from a retracted (pseudocryptorchid) one. Testicular descent during a course of hCG administration usually foretells permanent testicular descent at puberty, when circulating LH levels rise. Lack of descent usually means that orchiopexy will be necessary to preserve spermatogenesis.
Patients with constitutional delay in onset of puberty can be distinguished from those with hypogonadotropic hypogonadism using repeated hCG stimulation. Serum testosterone and estradiol levels rise in the former but not in the latter group.
Therapeutic Uses
As described above, hCG can be used in combination with hMG, uFSH, or rFSH to induce ovulation in women with hypogonadotropic hypogonadism or as part of an in vitro fertilization program. hCG stimulates testosterone secretion by the testes of men with hypogonadotropic hypogonadism. In such men, the increased intratesticular testosterone levels promote spermatogenesis, but FSH is often needed for fertility.
In patients with AIDS-related Kaposi's sarcoma, injection of hCG into the lesions has been reported to cause regression in a dose-related manner.
Dosage
The dosages for female and male infertility are described under hMG dosage. For prepubertal cryptorchidism, a dosage of 500–4000 units three times weekly for up to 6 weeks has been advocated.
Toxicity & Contraindications
Reported adverse effects include headache, depression, edema, precocious puberty, gynecomastia, or (rarely) production of antibodies to hCG. Human chorionic gonadotropin should be administered for infertility only by a physician with experience in this field. Androgen-dependent neoplasia and precocious puberty are contraindications to its use.
Prolactin
Prolactin is a 198-amino-acid peptide hormone produced in the anterior pituitary. Its structure resembles that of growth hormone. Prolactin is the principal hormone responsible for lactation. Milk production is stimulated by prolactin when appropriate circulating levels of estrogens, progestins, corticosteroids, and insulin are present. A deficiency of prolactin—which can occur in states of pituitary deficiency—is manifested by failure to lactate or by a luteal phase defect. In hypothalamic destruction, prolactin levels may be elevated as a result of impaired transport of prolactin-inhibiting hormone (dopamine) to the pituitary. Hyperprolactinemia can produce galactorrhea and hypogonadism and may be associated with symptoms of a pituitary mass. No preparation is available for use in prolactin-deficient patients. For patients with symptomatic hyperprolactinemia, inhibition of prolactin secretion can be achieved with cabergoline and other dopamine agonists.
Dopamine Agonists
Dopamine is released by the hypothalamus to inhibit prolactin release from the anterior pituitary. Bromocriptine, cabergoline, and pergolide are ergot derivatives with a very high affinity for dopamine D2 receptors in the pituitary. Quinagolide is a nonergot drug with similar D2 receptor affinity. These drugs lower circulating prolactin levels and shrink pituitary prolactin-secreting tumors. The chemical structure and pharmacokinetic features of bromocriptine are presented in Chapter 16: Histamine, Serotonin, & the Ergot Alkaloids.
Dopamine agonists decrease pituitary prolactin secretion through a dopamine-mimetic action on the pituitary at two central nervous system loci: (1) they decrease dopamine turnover in the tuberoinfundibular neurons of the arcuate nucleus, generating increased hypothalamic dopamine; and (2) they act directly on pituitary dopamine receptors to inhibit prolactin release.
These agents, like L-dopa, stimulate pituitary growth hormone release in normal subjects and—
paradoxically—suppress growth hormone release in acromegalics.
Pharmacokinetics
Cabergoline reaches peak plasma levels within 2–3 hours after a 1 mg oral dose. It has a half-life of 63–69 hours. Metabolites are excreted mostly in feces. Bromocriptine is metabolized more quickly.
All dopamine agonists may be administered orally. Additionally, bromocriptine and cabergoline are absorbed systemically after intravaginal insertion of tablets. Following intravaginal administration, serum levels peak more gradually.
Clinical Pharmacology
Prolactin-Secreting Adenomas
A dopamine agonist is the usual initial treatment for prolactinomas. Significant reduction in both tumor size and serum prolactin levels occurs in about 85% of those receiving these drugs for 6 months or longer.
Amenorrhea-Galactorrhea
Dopamine agonists are useful for treating problems induced by hyperprolactinemia: amenorrhea, galactorrhea, breast tenderness (mastodynia), infertility, and hypogonadism.
Physiologic Lactation
Dopamine agonists can prevent breast engorgement when breast feeding is not desired. Their use for this purpose has been discouraged because of toxicity (see below).
Acromegaly
A dopamine agonist alone or in combination with pituitary surgery, irradiation, or octreotide may be used to treat acromegaly. Acromegalic patients seldom respond adequately to bromocriptine unless the pituitary tumor secretes prolactin as well as growth hormone.
Parkinson's Disease and Restless Legs Syndrome
Cabergoline, bromocriptine, and pergolide have been used in Parkinson's disease to improve motor function and reduce levodopa requirements as discussed in Chapter 28: Pharmacologic Management of Parkinsonism & Other Movement Disorders. Cabergoline has also been effective in restless legs syndrome.
Preparations & Dosage
Cabergoline is initiated at 0.25 mg orally or vaginally twice weekly. It may be increased gradually according to serum prolactin determinations, up to a maximum of 1 mg twice weekly.
Bromocriptine is generally taken after the evening meal at the initial dose of 1.25 mg; the dose is then increased as tolerated. Most patients require 2.5–7.5 mg daily; acromegalics require higher doses, up to 20 mg/d. Bromocriptine tablets may be administered intravaginally to reduce nausea. Long-acting oral bromocriptine formulations (Parlodel SRO) and intramuscular formulations

(Parlodel L.A.R.) are available outside the USA.
Quinagolide (CV 205-502, Norprolac) in doses of 0.15–0.6 mg/d orally, suppresses prolactin and shrinks most prolactinomas. It also decreases cyclic mastodynia. Quinagolide is sometimes better tolerated than ergot-derived dopamine agonists. It is not available in the USA.
Toxicity & Contraindications
Dopamine agonists may cause nausea, headache, lightheadedness, orthostatic hypotension, and fatigue. Psychiatric manifestations occasionally occur even at lower doses and may take months to resolve. Erythromelalgia occurs rarely. High dosages of ergot-derived preparations may cause coldinduced peripheral digital vasospasm. Pulmonary infiltrates may occur with chronic high-dosage therapy. Cabergoline appears to cause nausea less often than bromocriptine. Vaginal administration of cabergoline or bromocriptine also tends to reduce nausea, but may cause vaginal irritation.
Dopamine agonist therapy during the early weeks of pregnancy has not been associated with an increased risk of spontaneous abortion or congenital malformations. Although there has been a longer experience with the safety of bromocriptine during early pregnancy, there is growing evidence that cabergoline is also safe to use in women with macroprolactinomas who must continue a dopamine agonist during pregnancy. In patients with small pituitary adenomas, dopamine agonist therapy is discontinued upon conception since there is usually no growth of microadenomas during pregnancy. Patients with very large adenomas require vigilance for tumor progression and often require a dopamine agonist throughout pregnancy. There have been rare reports of stroke or coronary thrombosis in postpartum women taking bromocriptine to suppress postpartum lactation.
If a woman who is receiving a dopamine agonist is late in having her menses, a pregnancy test is necessary; if she is amenorrheic, pregnancy tests should be performed regularly because ovulation may occur before menstruation resumes.
Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 37. Hypothalamic & Pituitary Hormones >
Posterior Pituitary Hormones
Two posterior pituitary hormones are known: vasopressin and oxytocin. Their structures are very similar. Posterior pituitary hormones are synthesized in the hypothalamus and then transported to the posterior pituitary, where they are stored and then released into the circulation.
Oxytocin
Oxytocin is a peptide hormone secreted by the posterior pituitary that elicits milk ejection in lactating women. It may contribute to the initiation of labor. Oxytocin is released during sexual orgasm.
Chemistry & Pharmacokinetics
Structure
Oxytocin is a nine-amino-acid peptide composed of a six-amino-acid disulfide ring and a threemembered tail (Figure 37–2). Oxytocin and vasopressin differ from vasotocin—the only posterior
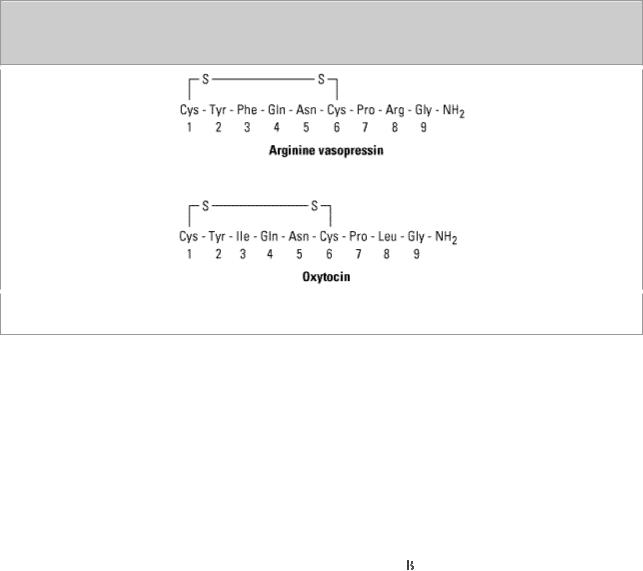
pituitary hormone found in nonmammalian vertebrates—by only one amino acid residue each.
Figure 37–2.
Posterior pituitary hormones. (Modified and reproduced, with permission, from Ganong WF:
Review of Medical Physiology, 21st ed. McGraw-Hill, 2003.)
Absorption, Metabolism, and Excretion
Oxytocin is usually administered intravenously for stimulation of labor. It is also available as a nasal spray to induce lactation postpartum. It is inactive if swallowed, because it is destroyed in the stomach and intestine. Oxytocin is not bound to plasma proteins and is catabolized by the kidneys and liver, with a circulating half-life of 5 minutes.
Pharmacodynamics
Oxytocin alters transmembrane ionic currents in myometrial smooth muscle cells to produce sustained uterine contraction. The sensitivity of the uterus to oxytocin increases during pregnancy. Oxytocin-induced myometrial contractions can be inhibited by  -adrenoceptor agonists, magnesium sulfate, or inhalation anesthetics. Oxytocin also causes contraction of myoepithelial cells surrounding mammary alveoli, which leads to milk ejection. Without oxytocin-induced contraction, normal lactation cannot occur. Oxytocin has weak antidiuretic and pressor activity.
-adrenoceptor agonists, magnesium sulfate, or inhalation anesthetics. Oxytocin also causes contraction of myoepithelial cells surrounding mammary alveoli, which leads to milk ejection. Without oxytocin-induced contraction, normal lactation cannot occur. Oxytocin has weak antidiuretic and pressor activity.
Clinical Pharmacology
Diagnostic Uses
Oxytocin infusion near term will produce uterine contractions that decrease the fetal blood supply. The fetal heart rate response to a standardized oxytocin challenge test provides information about placental circulatory reserve. An abnormal response suggests intrauterine growth retardation and may warrant immediate cesarean delivery.
Therapeutic Uses
Oxytocin is used to induce labor and augment dysfunctional labor for (1) conditions requiring early vaginal delivery (eg, Rh problems, maternal diabetes, or preeclampsia), (2) uterine inertia, and (3) incomplete abortion. Oxytocin can also be used for control of postpartum uterine hemorrhage.
Impaired milk ejection may respond to nasal oxytocin. Synthetic peptide and nonpeptide oxytocin antagonists that can prevent premature labor are being investigated.
Dosage
Oxytocin is frequently given to induce and maintain labor after the cervix has ripened naturally or with the aid of misoprostol. For induction of labor, oxytocin should be administered intravenously via an infusion pump with appropriate fetal and maternal monitoring. An initial infusion rate of 1 mU/min is increased every 15–30 minutes until a physiologic contraction pattern is established. The maximum infusion rate is 20 mU/min. For postpartum uterine bleeding, 10–40 units is added to 1 L of 5% dextrose, and the infusion rate is titrated to control uterine atony. Alternatively, 10 units can be given intramuscularly after delivery of the placenta. To induce milk let-down, one puff is sprayed into each nostril in the sitting position 2–3 minutes before nursing.
Toxicity & Contraindications
When oxytocin is used properly, serious toxicity is rare. Among the reported adverse reactions are maternal deaths due to hypertensive episodes, uterine rupture, water intoxication, and fetal deaths. Afibrinogenemia has also been reported.
Contraindications include fetal distress, prematurity, abnormal fetal presentation, cephalopelvic disproportion, and other predispositions for uterine rupture.
Vasopressin (Antidiuretic Hormone, ADH)
Vasopressin is a peptide hormone released by the posterior pituitary in response to rising plasma tonicity or falling blood pressure. Vasopressin possesses antidiuretic and vasopressor properties. A deficiency of this hormone results in diabetes insipidus (see Chapters 15 and 17).
Chemistry & Pharmacokinetics
Structure
Vasopressin is a nonapeptide with a six-amino-acid ring and a three-amino-acid side chain. The residue at position 8 is arginine in humans and in most other mammals except pigs and related species, whose vasopressin contains lysine at position 8 (Figure 37–2).
Absorption, Metabolism, and Excretion
Vasopressin is administered by intravenous, intramuscular, or intranasal routes; oral absorption is slight. The half-life of circulating ADH is approximately 20 minutes, with renal and hepatic catabolism via reduction of the disulfide bond and peptide cleavage. A small amount of vasopressin is excreted as such in the urine.
Pharmacodynamics
Vasopressin interacts with two types of receptors. V1 receptors are found on vascular smooth muscle cells and mediate vasoconstriction (see Chapter 17: Vasoactive Peptides). V2 receptors are found on renal tubule cells and mediate antidiuresis through increased water permeability and water resorption in the collecting tubules. Extrarenal V2-like receptors mediate release of coagulation


 g (0.1–0.2 mL) intranasally at bedtime, is used.
g (0.1–0.2 mL) intranasally at bedtime, is used.
Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 37. Hypothalamic & Pituitary Hormones >
Preparations Available
Bromocriptine (Parlodel)
Oral: 2.5 mg tablets, 5 mg capsules
Cabergoline (Dostinex)
Oral: 0.5 mg scored tablets
Cetrorelix (Cetrotide)
Parenteral: 0.25, 3.0 mg/vial with diluent for subcutaneous injection
Chorionic gonadotropin [hCG] (generic, Profasi, A.P.L., Pregnyl, others) Parenteral: powder to reconstitute 500, 1000, 2000 units/mL for injection
Corticorelin ovine (Acthrel)
Parenteral: 100  g for IV injection
g for IV injection
Corticotropin (H.P. Acthar Gel)
Parenteral: 80 units/mL
Cosyntropin (Cortrosyn)
Parenteral: 0.25 mg/vial with diluent for IV or IM injection
Desmopressin (DDAVP, Stimate)
Nasal: 0.1, 1.5 mg/mL solution
Nasal: 100 g/mL spray pump and rhinal tube delivery system
Parenteral: 4 g/mL solution for injection
Oral: 0.1, 0.2 mg tablets
Follitropin alfa (Gonal-F)
Parenteral: 37.5, 150 IU powder for injection
Follitropin beta [FSH] (Follistim)
Parenteral: 75 IU powder for injection

Ganirelix (Antagon)
Parenteral: 500  g/mL for injection
g/mL for injection
Gonadorelin acetate [GnRH] (Lutrepulse)
Parenteral: powder to reconstitute for injection via Lutrepulse pump (0.8, 3.2 mg/vial)
Gonadorelin hydrochloride [GnRH] (Factrel)
Parenteral: 100, 500 mg for injection
Goserelin acetate (Zoladex)
Parenteral: 3.6, 10.8 mg subcutaneous implant
Histrelin (Supprelin)
Parenteral: 120, 300, 600 mg for subcutaneous injection
Leuprolide (generic, Lupron)
Parenteral: 5 mg/mL for subcutaneous injection
Parenteral depot suspension (Lupron Depot, Depot-Ped, Depot-3, Depot-4): lyophilized microspheres to reconstitute for IM injection (3.75, 7.5, 11.25, 15, 22.5, 30 mg/vial)
Parenteral implant: 72 mg
Menotropins [hMG] (Pergonal, Repronex)
Parenteral: 75 IU FSH and 75 IU LH activity, 150 IU FSH and 150 IU LH activity, each with diluent
Nafarelin (Synarel)
Nasal: 2 mg/mL (200 g/spray)
Octreotide (Sandostatin)
Parenteral: 0.05, 0.1, 0.2, 0.5, 1.0 mg/mL for subcutaneous or IV administration
Parenteral depot injection (Sandostatin LAR Depot): 10, 20, 30 mg for IM injection only
Oxytocin (generic, Pitocin, Syntocinon)
Parenteral: 10 units/mL for injection
Nasal: 40 units/mL spray

Pergolide (Permax)
Oral: 0.05, 0.25, 1.0 mg tablets
Protirelin (Thypinone, Relefact TRH, Thyrel TRH)
Parenteral: 500 mg/mL for injection
Sermorelin (Geref)
Parenteral: 0.5, 1.0 mg for subcutaneous injection; 50 
 g powder to reconstitute for intravenous injection
g powder to reconstitute for intravenous injection
Somatrem (Protropin)
Parenteral: 5, 10 mg/vial with diluent for subcutaneous or IM injection
Somatropin (Genotropin, Humatrope, Nutropin, Nutropin AQ, Norditropin, Serostim, Saizen)
Parenteral: 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.5, 1.6, 1.8, 2, 4, 5, 5.8, 6, 8, 10, 12, 13.5, 13.8, 15, 18, 22.5, 24 mg/vial with diluent for subcutaneous or IM injection
Thyrotropin alpha (Thyrogen)
Parenteral: 1.1 mg (> 4 IU)/vial with diluent for IM injection
Triptorelin (Trelstar)
Parenteral: 3.75, 11.25 mg for IM injection
Urofollitropin (Fertinex, Bravelle)
Parenteral: powder to reconstitute for injection, 75, 150 IU FSH activity per ampule
Vasopressin (generic, Pitressin)
Parenteral: 20 pressor units/mL for IM or subcutaneous administration
Chapter 38. Thyroid & Antithyroid Drugs
Katzung PHARMACOLOGY, 9e > Section VII. Endocrine Drugs > Chapter 38. Thyroid & Antithyroid Drugs >
Thyroid & Antithyroid Drugs: Introduction
Because of its anatomic prominence, the thyroid was one of the first of the endocrine glands to be associated with the clinical conditions caused by its malfunction.
Thyroid Physiology