

2. Фенилкетонурия. Причина этой наследственной патологии, покажите «блок» фермента на схеме. Проявления.
Фенилкетонурия
В печени здоровых людей небольшая часть фенилаланина (∼10%) превращается в фенил-лактат и фенилацетилглутамин (рис. 9-30). Этот путь катаболизма фенилаланина становится главным при нарушении основного пути - превращения в тирозин, катализируемого фенил-аланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглу-тамина. Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ:
Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20-30 раз (в норме - 1,0-2,0 мг/дл), в моче - в 100-300 раз по сравнению с нормой (30 мг/дл). Концентрация фенилпирувата и фениллактата в моче достигает 300-600 мг/дл при полном отсутствии в норме.
Наиболее тяжёлые проявления ФКУ - нарушение умственного и физического развития, судорожный синдром, нарушение пигментации. При отсутствии лечения больные не доживают до 30 лет. Частота заболевания - 1:10 000 новорождённых. Заболевание наследуется по аутосомно-рецессивному типу.
Тяжёлые проявления ФКУ связаны с токсическим действием на клетки мозга высоких концентраций фенилаланина, фенилпирувата, фениллактата. Большие концентрации фенилаланина ограничивают транспорт тирозина и триптофана через гематоэнцефаличеекий барьер и тормозят синтез нейро-медиаторов (дофамина, норадреналина, серотонина).
Вариантная ФКУ (коферментзависимая гиперфенилаланинемия) - следствие мутаций в генах, контролирующих метаболизм Н4БП. Клинические проявления - близкие, но не точно совпадающие с проявлениями классической ФКУ. Частота заболевания - 1-2 случая на 1 млн новорождённых.
Н4БП необходим для реакций гидроксилирования не только фенилаланина, но также тирозина и триптофана, поэтому при недостатке этого кофермента нарушается метаболизм всех 3 аминокислот, в том числе и синтез ней-ромедиаторов. Заболевание характеризуется тяжёлыми неврологическими нарушениями и ранней смертью ("злокачественная" ФКУ).
Прогрессирующее нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина. Если такое лечение начато сразу после рождения ребёнка, то повреждение мозга предотвращается. Считается, что ограничения в питании могут быть ослаблены после 10-летнего возраста (окончание процессов миелиниза-ции мозга), однако в настоящее время многие педиатры склоняются в сторону "пожизненной диеты".
Для диагностики ФКУ используют качественные и количественные методы обнаружения патологических метаболитов в моче, определение концентрации фенилаланина в крови и моче. Дефектный ген, ответственный за фенилкетонурию, можно обнаружить у фенотипически нормальных гетерозиготных носителей с помощью теста толерантности к фенилаланину. Для этого обследуемому дают натощак ∼10 г фенилаланина в виде раствора, затем через часовые интервалы берут пробы крови, в которых определяют содержание тирозина. В норме концентрация тирозина в крови после фенилаланиновой нагрузки значительно выше, чем у гетерозиготных носителей гена фежилкетонурии. Этот тест используется в генетической консультации для определения риска рождения больного ребёнка. Разработана схема скрининга для выявления новорождённых детей с ФКУ. Чувствительность теста практически достигает 100%.
В настоящее время диагностику мутантного гена, ответственного за ФКУ, можно проводить с помощью методов ДНК-диагностики (рестрикционного анализа и ПЦР).
Рис.
9-30. Альтернативные пути катаболизма
фенилаланина. При
дефекте фенилаланингидроксилазы
накопившийся фенилалан и н подвергается
трансаминированию с а-кетоглутаратом.
Образовавшийся фенилпируват превращается
либо в фениллактат, либо в фенилацетилглутамин,
которые накапливаются в крови и выделяются
с мочой. Эти соединения токсичны для
клеток мозга.
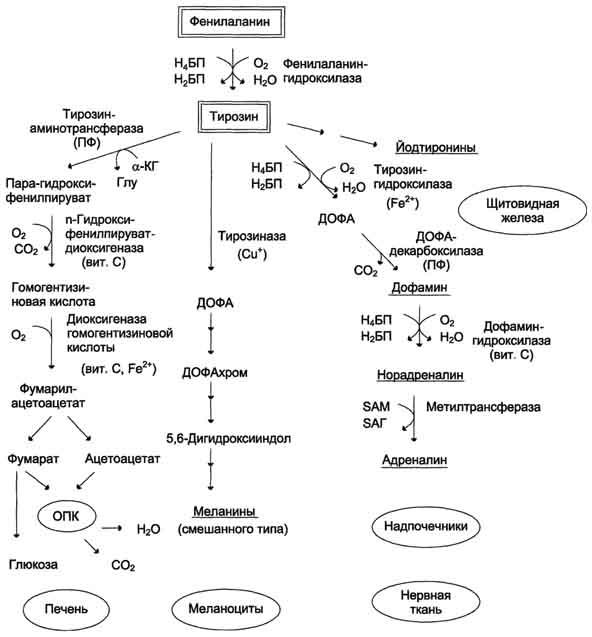
Рис. 9-28. Пути превращения фенилаланина и тирозина в разных тканях. Н4БП - тетрагидробиоптерин; Н2БП - дигидробиоптерин; ПФ - пиридоксальфосфат; SAM - S-аденозилметионин.